

Systematic errors in orthology inference and their effects on evolutionary analyses
- PMID: 33659875
- PMCID: PMC7892920
- DOI: 10.1016/j.isci.2021.102110
Systematic errors in orthology inference and their effects on evolutionary analyses
Abstract
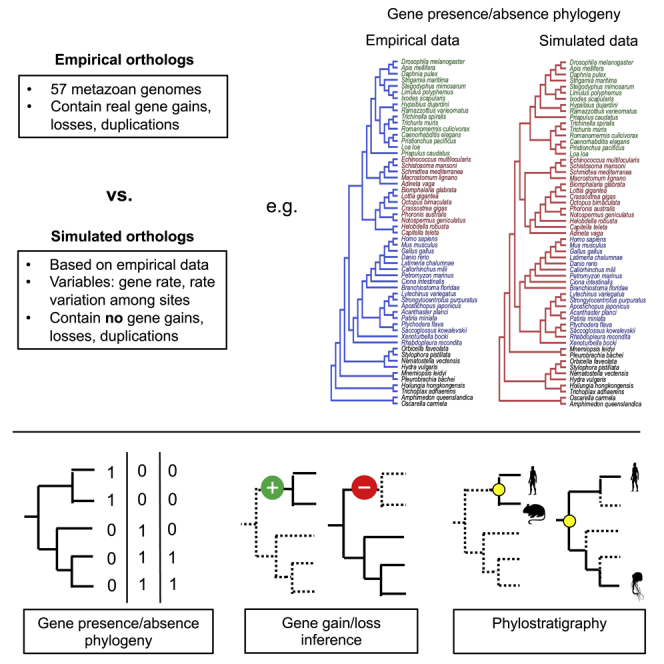
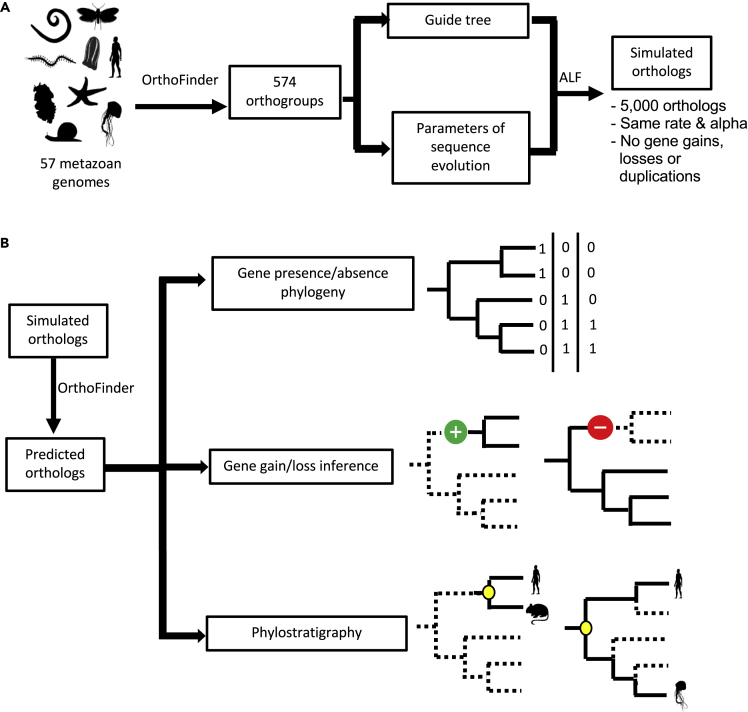
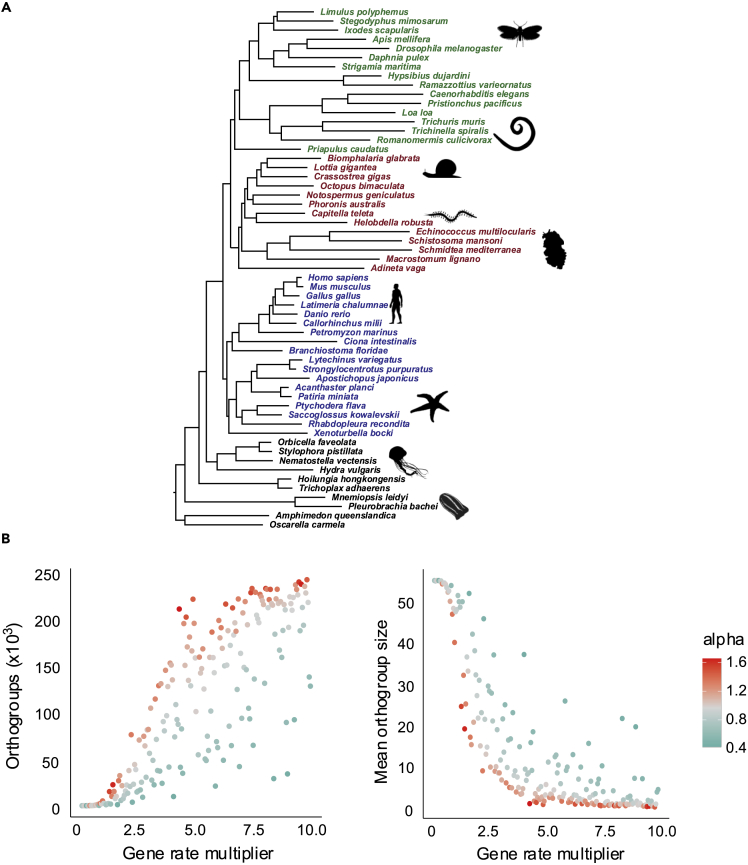
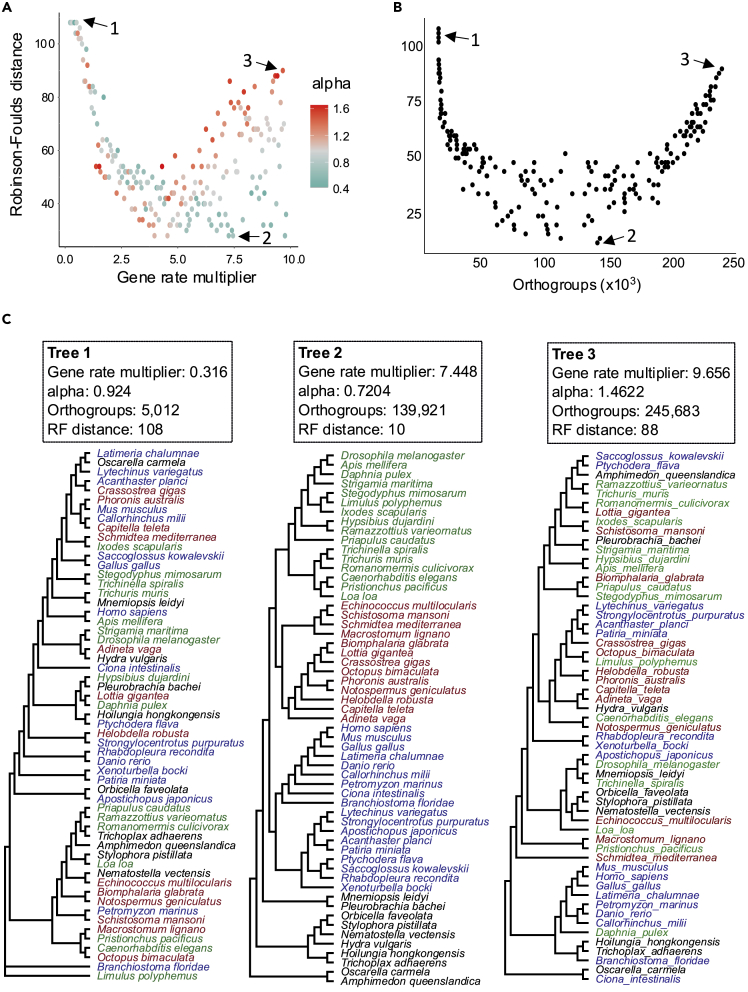
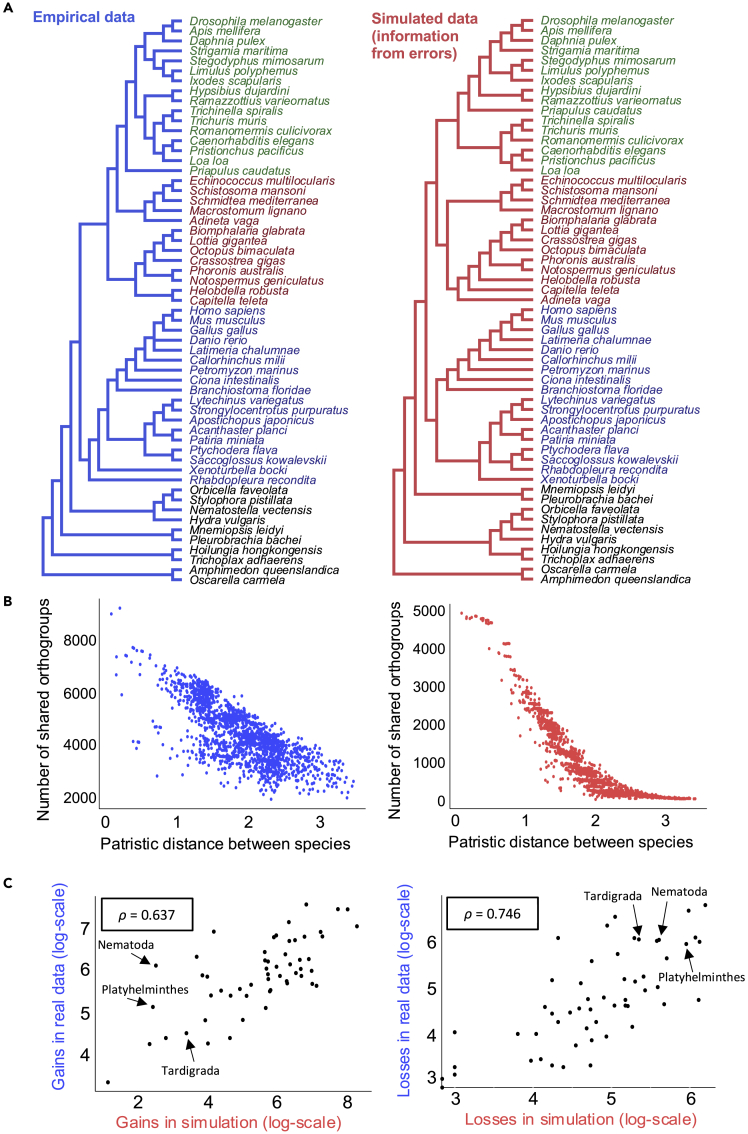
The availability of complete sets of genes from many organisms makes it possible to identify genes unique to (or lost from) certain clades. This information is used to reconstruct phylogenetic trees; identify genes involved in the evolution of clade specific novelties; and for phylostratigraphy-identifying ages of genes in a given species. These investigations rely on accurately predicted orthologs. Here we use simulation to produce sets of orthologs that experience no gains or losses. We show that errors in identifying orthologs increase with higher rates of evolution. We use the predicted sets of orthologs, with errors, to reconstruct phylogenetic trees; to count gains and losses; and for phylostratigraphy. Our simulated data, containing information only from errors in orthology prediction, closely recapitulate findings from empirical data. We suggest published downstream analyses must be informed to a large extent by errors in orthology prediction that mimic expected patterns of gene evolution.
Keywords: Biological Sciences; Evolutionary Biology; Evolutionary Mechanisms; Evolutionary Processes; Phylogenetics; Phylogeny.
© 2021 The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Altenhoff A.M., Glover N.M., Dessimoz C. Inferring orthology and paralogy. In: Anisimova M., editor. Evolutionary Genomics. Springer; 2019.
-
- Buchfink B., Xie C., Huson D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 2015;12:59–60. - PubMed
-
- Cannon J.T., Vellutini B.C., Smith J., Ronquist F., Jondelius U., Hejnol A. Xenacoelomorpha is the sister group to Nephrozoa. Nature. 2016;530:89–93. - PubMed
-
- Domazet-Lošo T., Brajković J., Tautz D. A phylostratigraphy approach to uncover the genomic history of major adaptations in metazoan lineages. Trends Genet. 2007;23:533–539. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
