

Context-Dependent Glioblastoma-Macrophage/Microglia Symbiosis and Associated Mechanisms
- PMID: 33663953
- PMCID: PMC8005482
- DOI: 10.1016/j.it.2021.02.004
Context-Dependent Glioblastoma-Macrophage/Microglia Symbiosis and Associated Mechanisms
Abstract
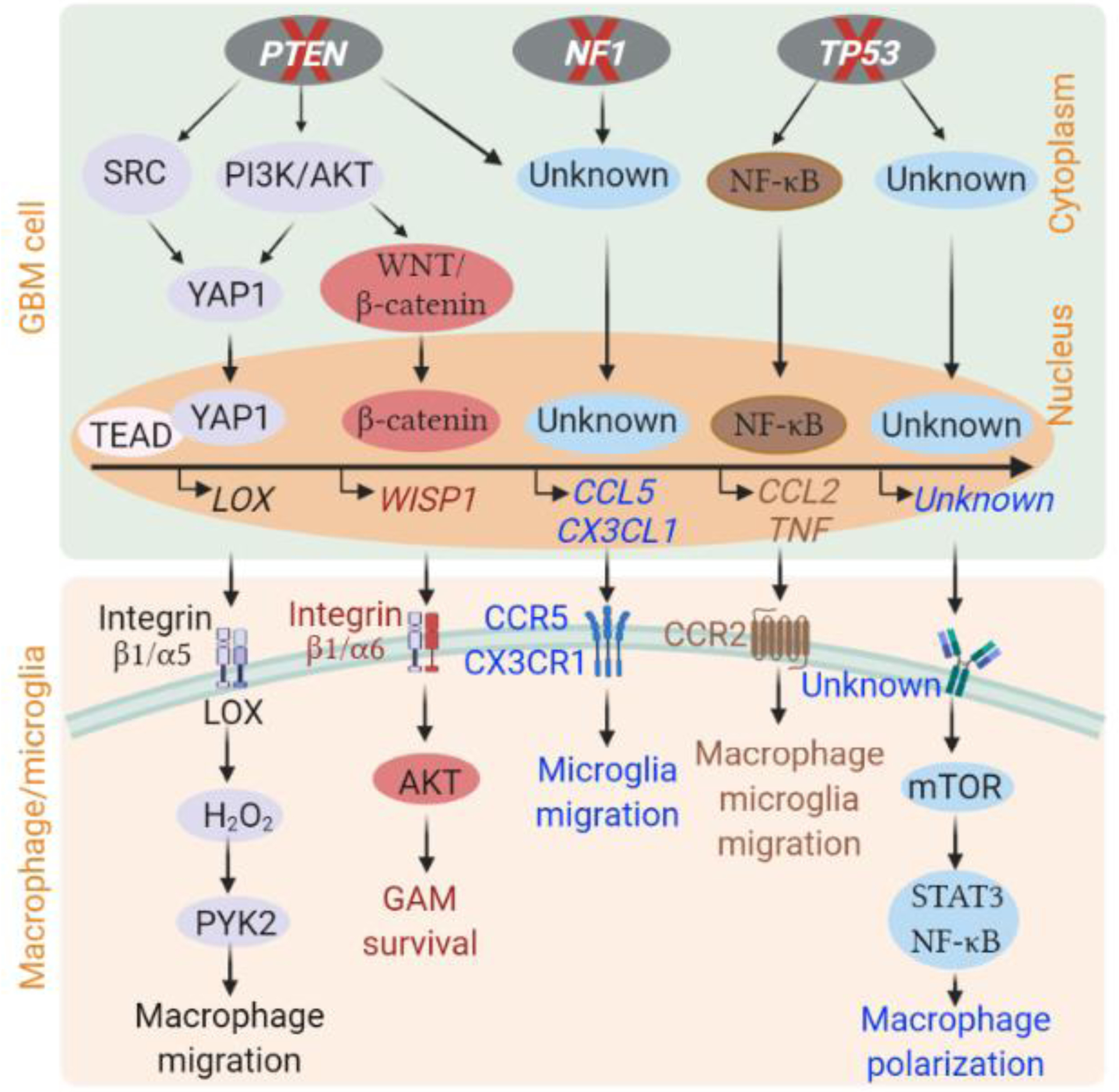
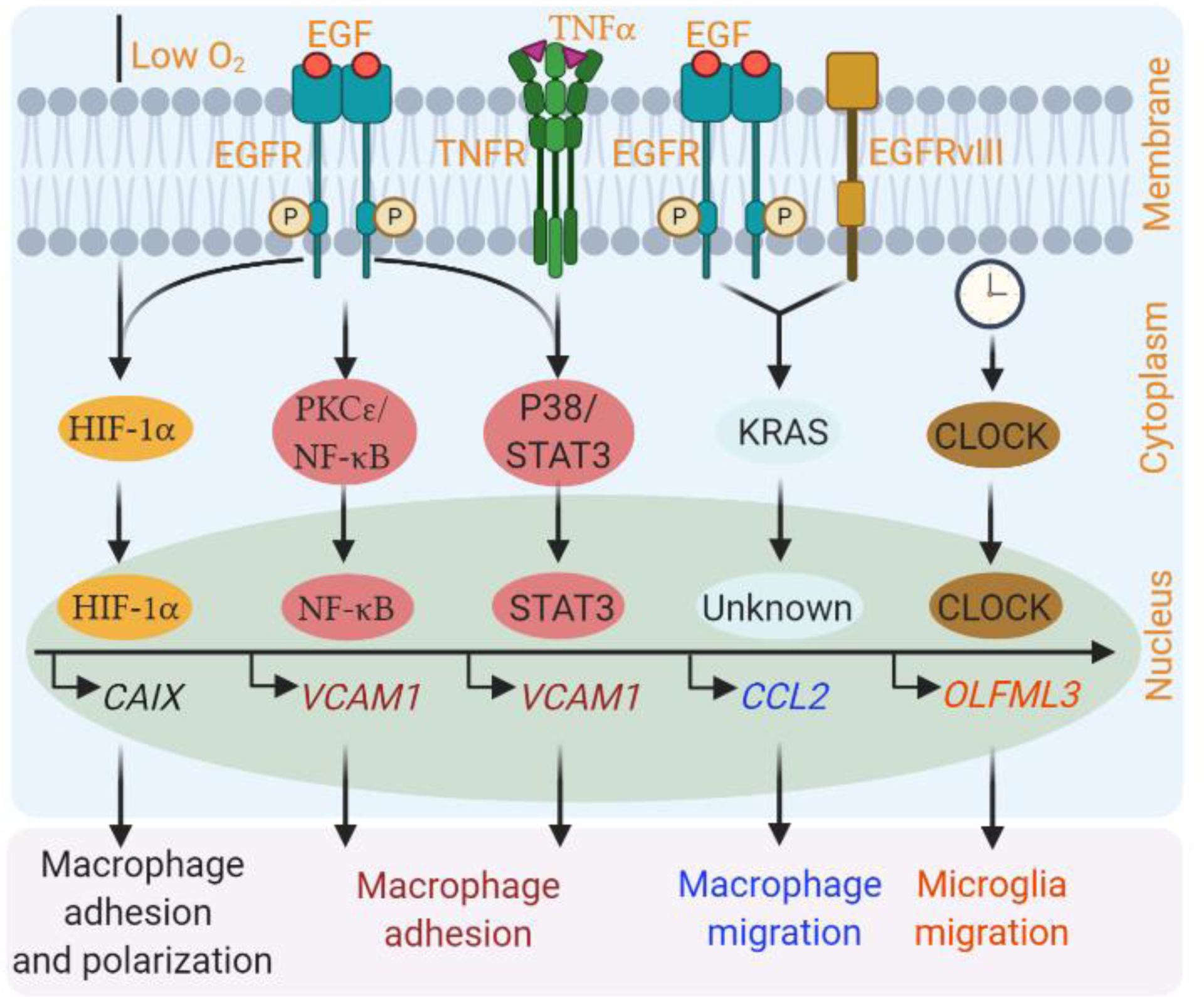
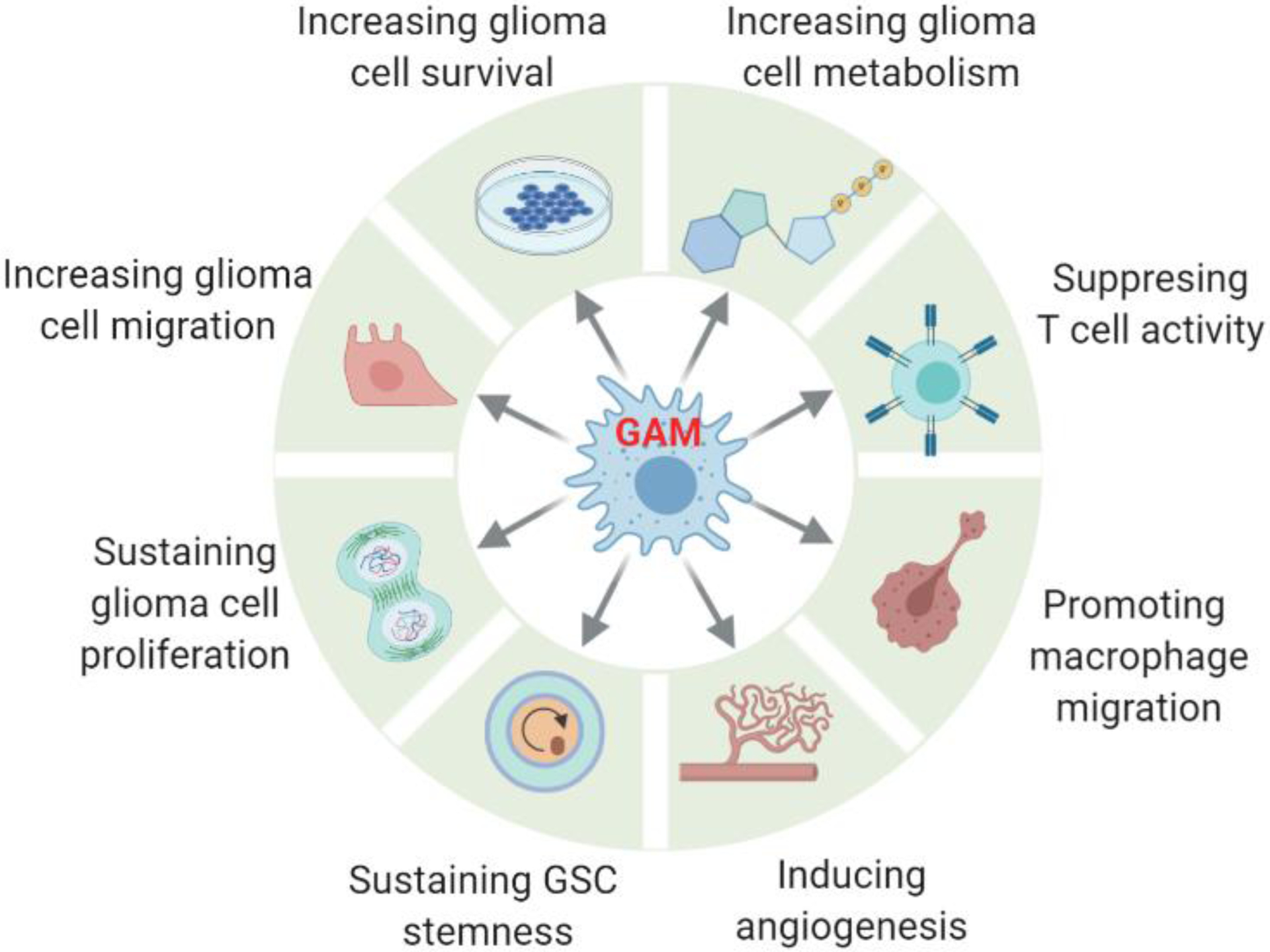
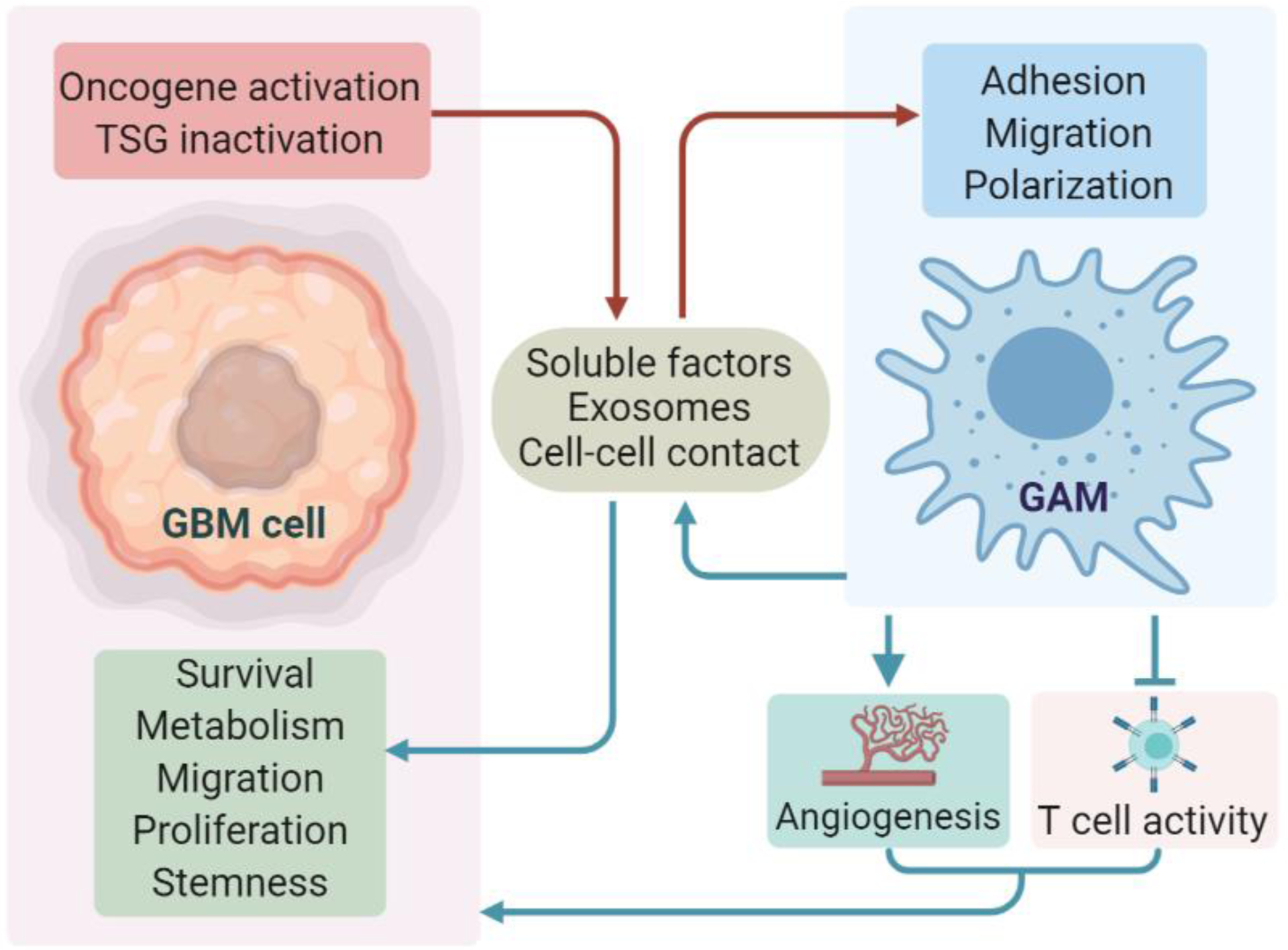
Glioblastoma (GBM) is a lethal form of primary brain tumor in human adults. The impact of tumor-intrinsic alterations is not exclusively confined to cancer cells but can also be extended to the tumor microenvironment (TME). Glioblastoma-associated macrophages/microglia (GAMs) are a prominent type of immune cells that account for up to 50% of total cells in GBM. Emerging evidence suggests that context-dependent GBM-GAM symbiotic interactions are pivotal for tumor growth and progression. Here, we discuss how specific genetic alterations in GBM cells affect GAM biology and, reciprocally, how GAMs support GBM progression. We hypothesize that understanding context-dependent GBM-GAM symbiosis may reveal the molecular basis of GBM tumorigenesis and lead to novel candidate treatment approaches aiming to improve GBM patient outcomes.
Keywords: crosstalk; glioblastoma; heterogeneity; macrophages; microglia; symbiosis.
Copyright © 2021 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of Interests No interests are declared.
Figures
References
-
- Wellenstein MD and de Visser KE (2018) Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 48 (3), 399–416. - PubMed
-
- Geraldo LHM et al. (2019) Glioblastoma Therapy in the Age of Molecular Medicine. Trends Cancer 5 (1), 46–65. - PubMed
-
- Zanders ED et al. (2019) Therapy for glioblastoma: is it working? Drug Discov Today 24 (5), 1193–1201. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
