

Identification of rare and common regulatory variants in pluripotent cells using population-scale transcriptomics
- PMID: 33664507
- PMCID: PMC7944648
- DOI: 10.1038/s41588-021-00800-7
Identification of rare and common regulatory variants in pluripotent cells using population-scale transcriptomics
Abstract
Induced pluripotent stem cells (iPSCs) are an established cellular system to study the impact of genetic variants in derived cell types and developmental contexts. However, in their pluripotent state, the disease impact of genetic variants is less well known. Here, we integrate data from 1,367 human iPSC lines to comprehensively map common and rare regulatory variants in human pluripotent cells. Using this population-scale resource, we report hundreds of new colocalization events for human traits specific to iPSCs, and find increased power to identify rare regulatory variants compared with somatic tissues. Finally, we demonstrate how iPSCs enable the identification of causal genes for rare diseases.
Conflict of interest statement
Conflicts of interest
S.B.M. is on SAB of Myome Inc.
Figures
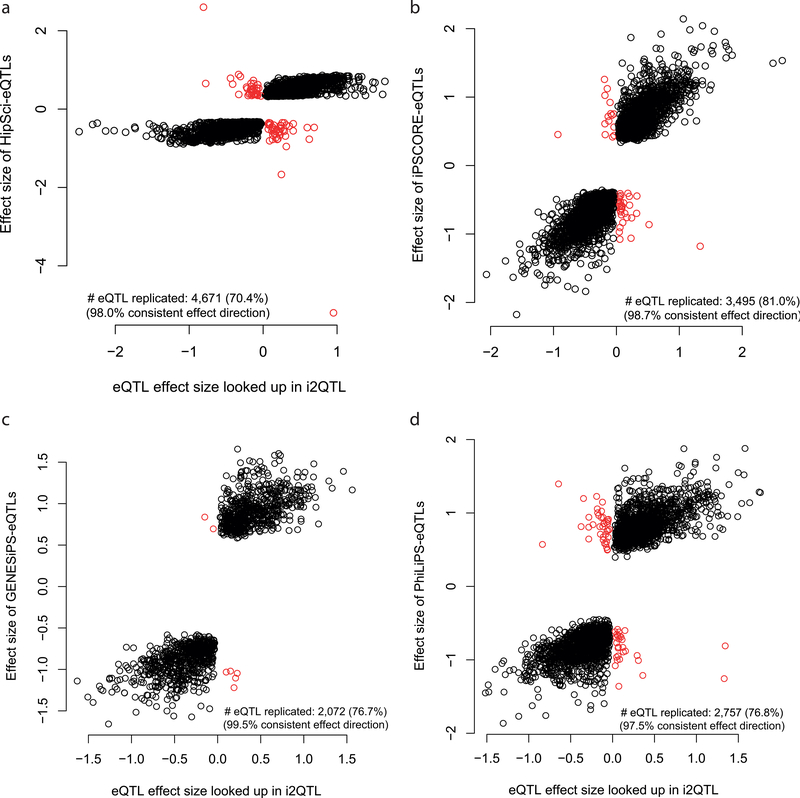
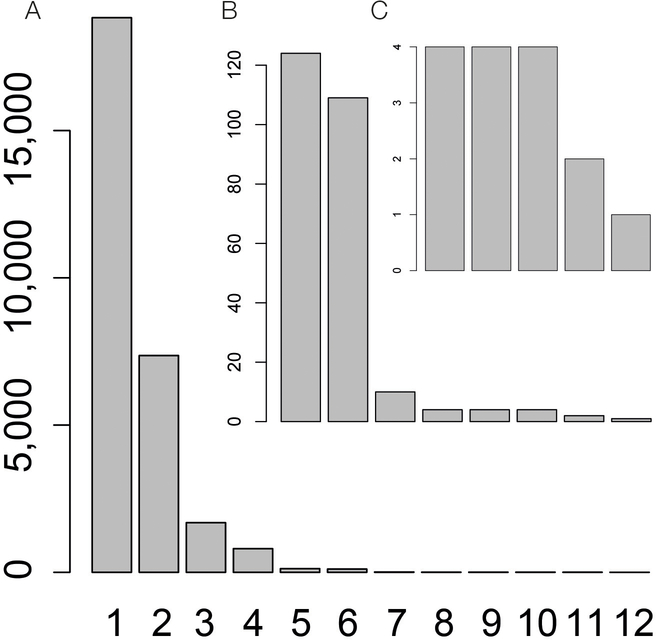
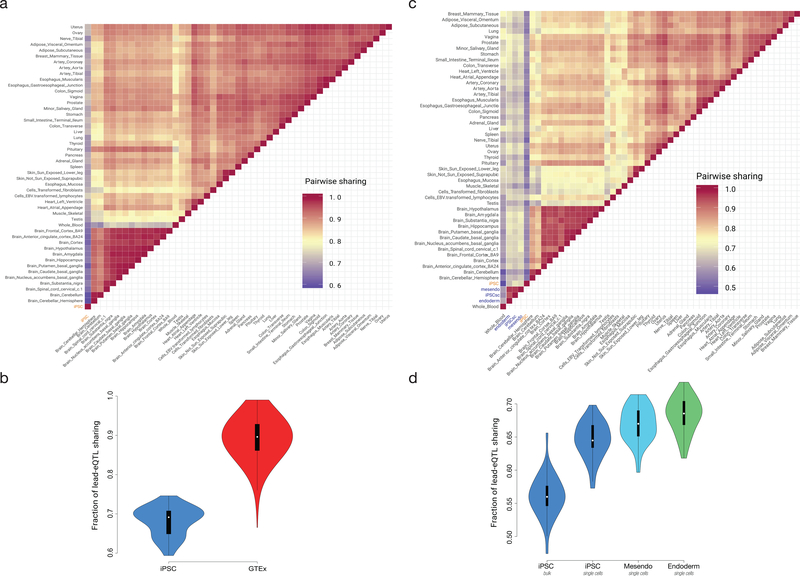
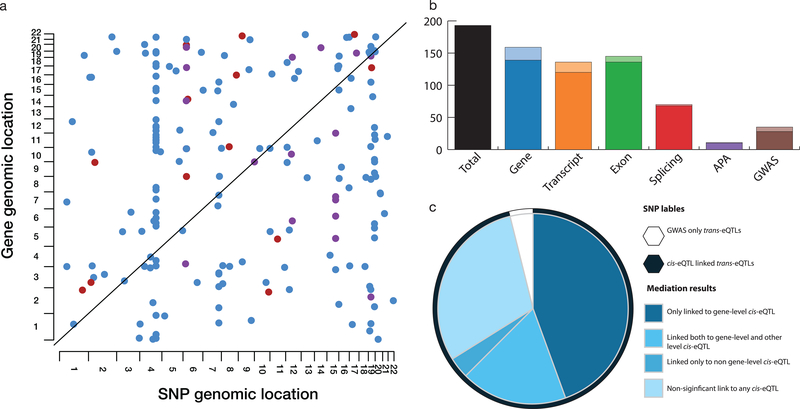
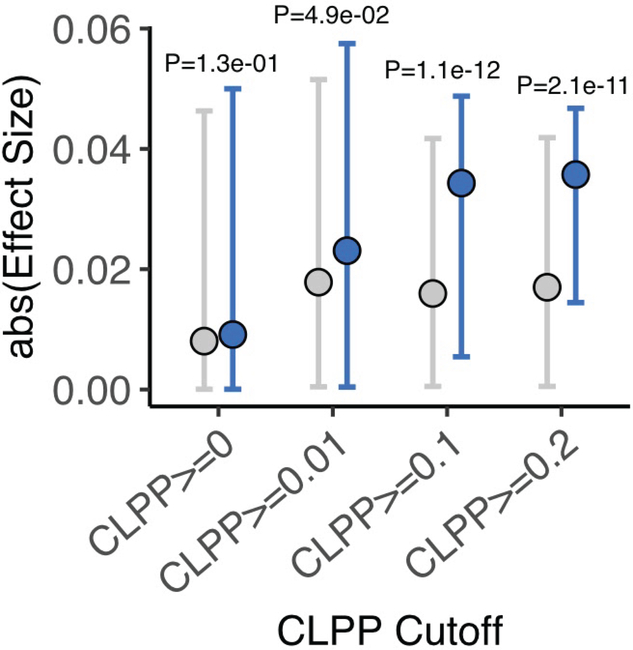
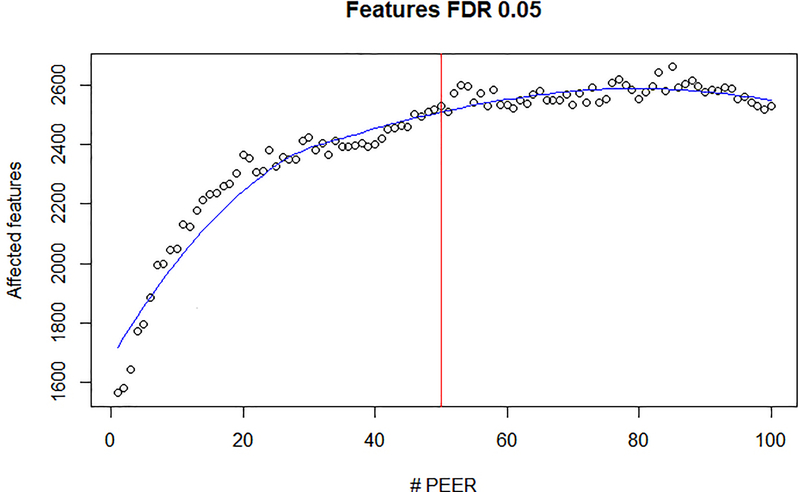
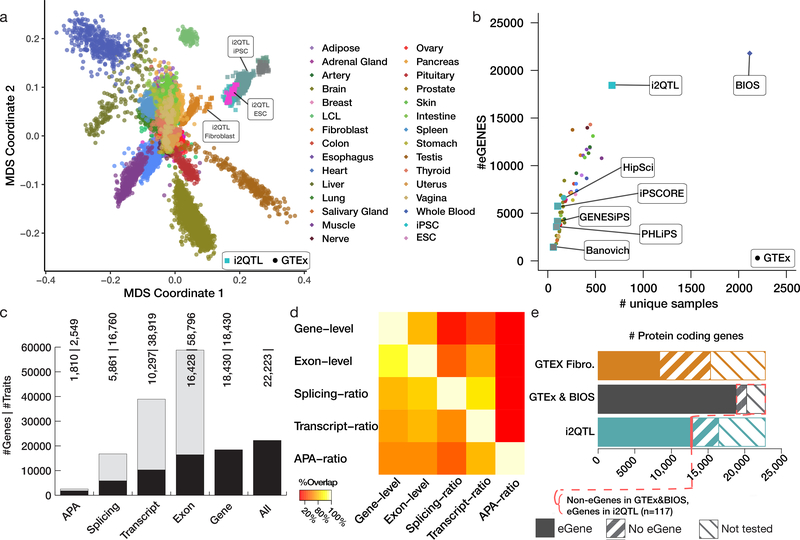
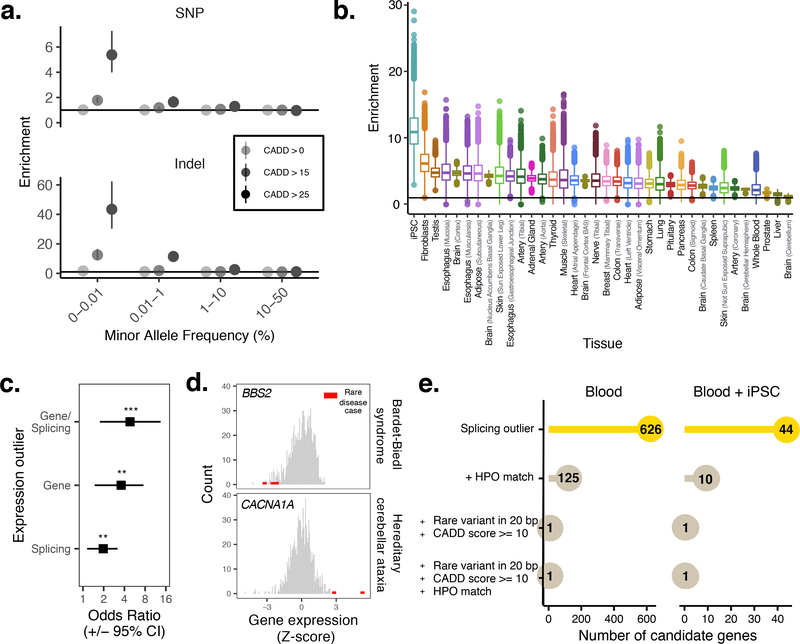
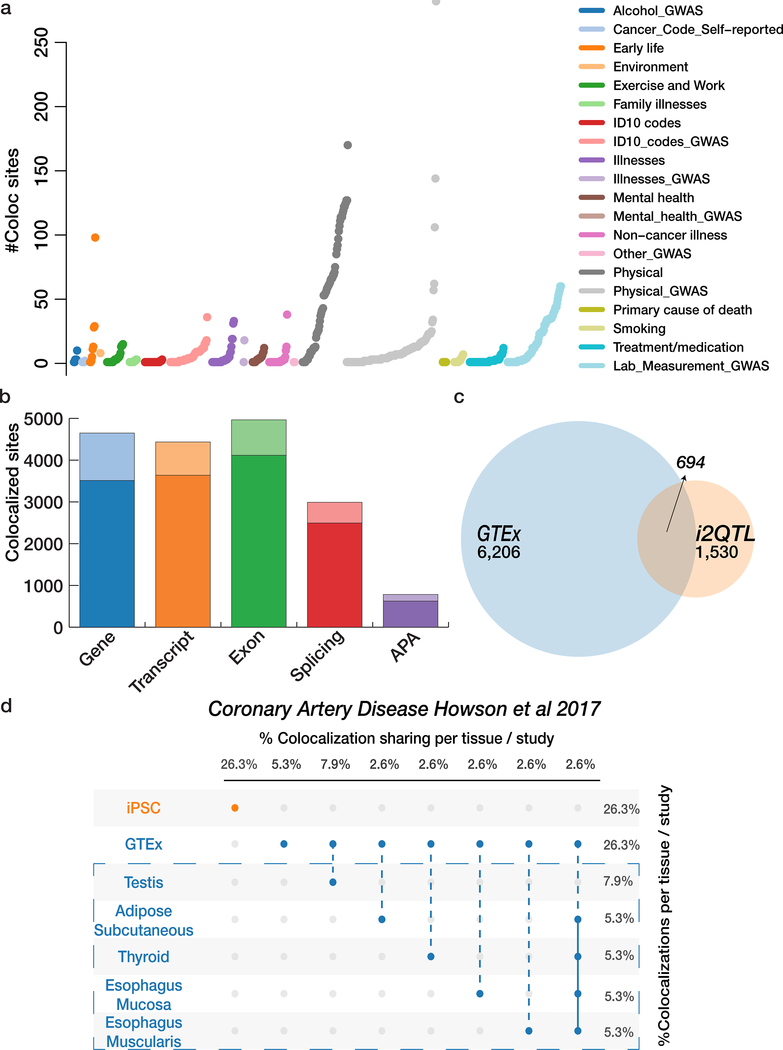
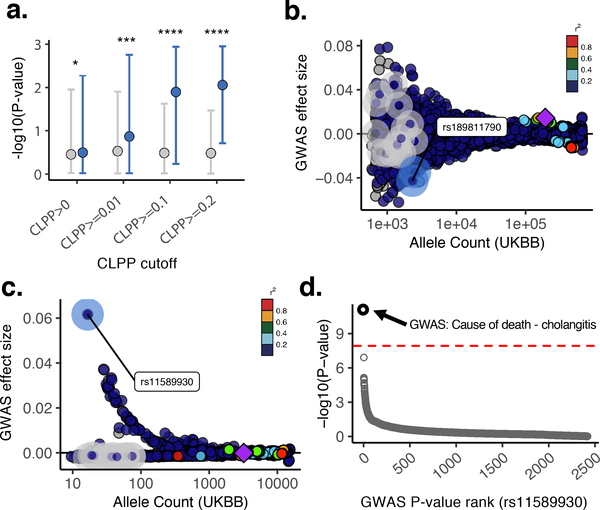
Comment in
-
Stem cells root out genetic variants.Nat Rev Genet. 2021 May;22(5):265. doi: 10.1038/s41576-021-00357-5. Nat Rev Genet. 2021. PMID: 33785895 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- U01 DK105541/DK/NIDDK NIH HHS/United States
- DP3 DK112155/DK/NIDDK NIH HHS/United States
- U01 HG007708/HG/NHGRI NIH HHS/United States
- R01 DK107437/DK/NIDDK NIH HHS/United States
- U01 HG010218/HG/NHGRI NIH HHS/United States
- R01 HL142015/HL/NHLBI NIH HHS/United States
- U01 HG009080/HG/NHGRI NIH HHS/United States
- R01 DK116750/DK/NIDDK NIH HHS/United States
- R01 AG066490/AG/NIA NIH HHS/United States
- P30 DK116074/DK/NIDDK NIH HHS/United States
- U01 HL107388/HL/NHLBI NIH HHS/United States
- T32 LM012409/LM/NLM NIH HHS/United States
- WT098503/WT_/Wellcome Trust/United Kingdom
- U01 HG009431/HG/NHGRI NIH HHS/United States
- R01 HG008150/HG/NHGRI NIH HHS/United States
- R01 DK106236/DK/NIDDK NIH HHS/United States
- U01 HL107442/HL/NHLBI NIH HHS/United States
- R01 DK120565/DK/NIDDK NIH HHS/United States
- T15 LM007033/LM/NLM NIH HHS/United States
- MRC_/Medical Research Council/United Kingdom
- WT090851/WT_/Wellcome Trust/United Kingdom
- S10 OD023452/OD/NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
