

The lncRNA Malat1 regulates microvascular function after myocardial infarction in mice via miR-26b-5p/Mfn1 axis-mediated mitochondrial dynamics
- PMID: 33667993
- PMCID: PMC7937833
- DOI: 10.1016/j.redox.2021.101910
The lncRNA Malat1 regulates microvascular function after myocardial infarction in mice via miR-26b-5p/Mfn1 axis-mediated mitochondrial dynamics
Abstract
Rationale: Myocardial infarction (MI) is a leading cause of cardiovascular mortality globally. The improvement of microvascular function is critical for cardiac repair after MI. Evidence now points to long non-coding RNAs (lncRNAs) as key regulators of cardiac remodelling processes. The lncRNA Malat1 is involved in the development and progression of multiple cardiac diseases. Studies have shown that Malat1 is closely related to the regulation of endothelial cell regeneration. However, the potential molecular mechanisms of Malat1 in repairing cardiac microvascular dysfunction after MI remain unreported.
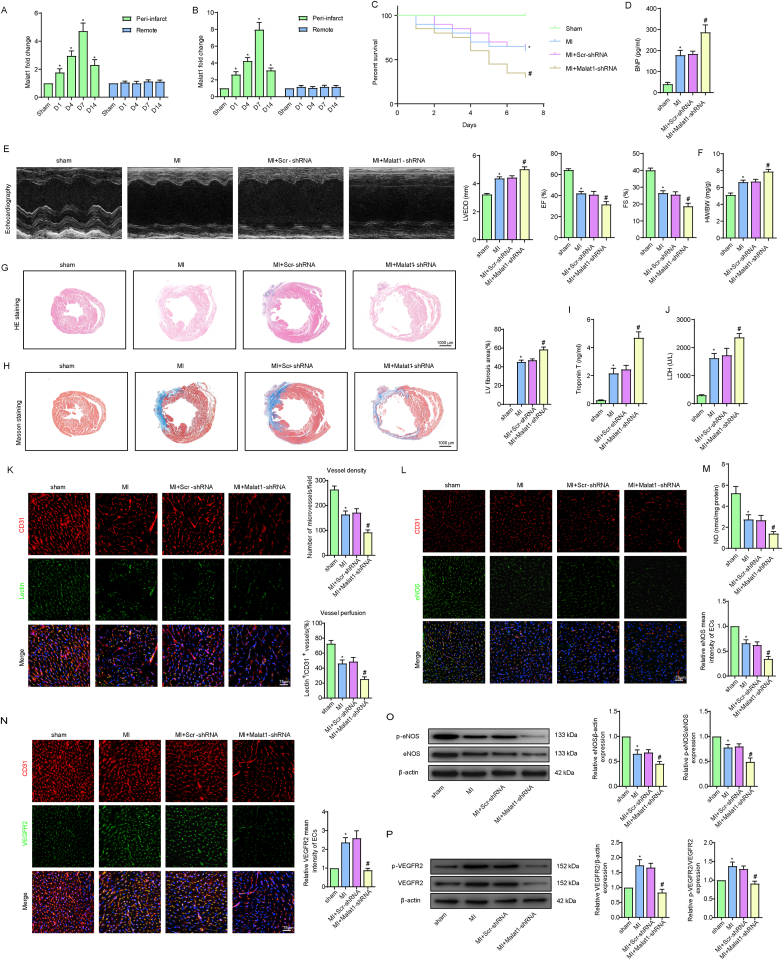
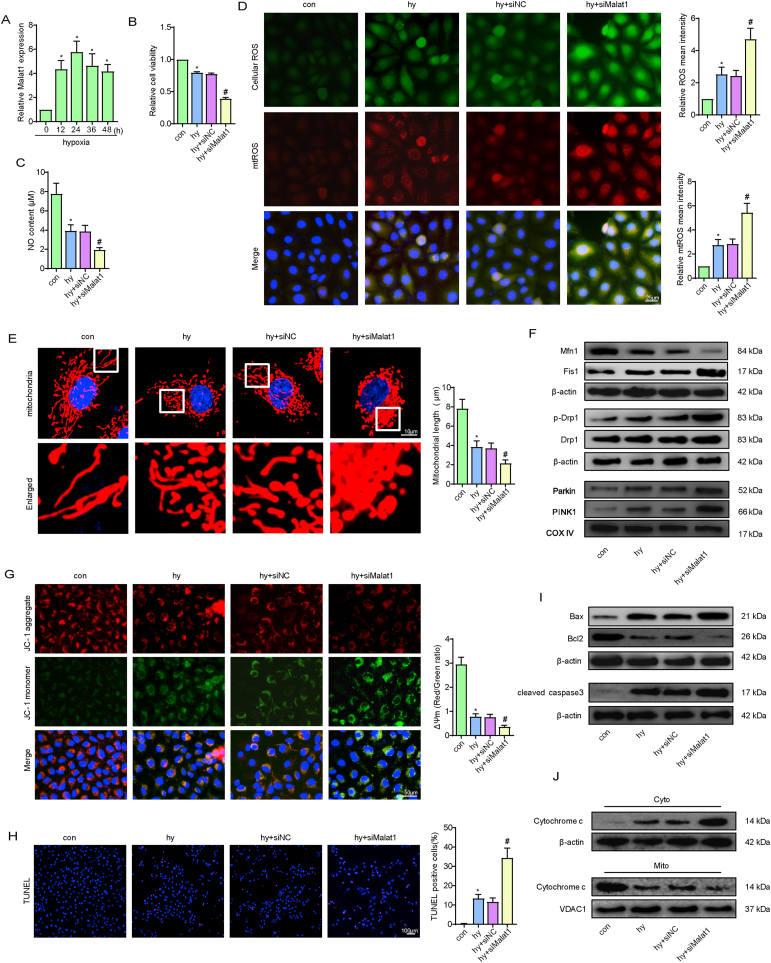
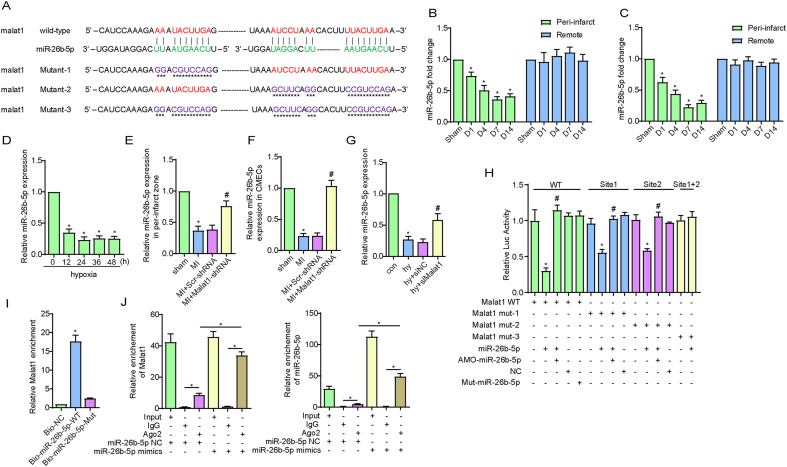
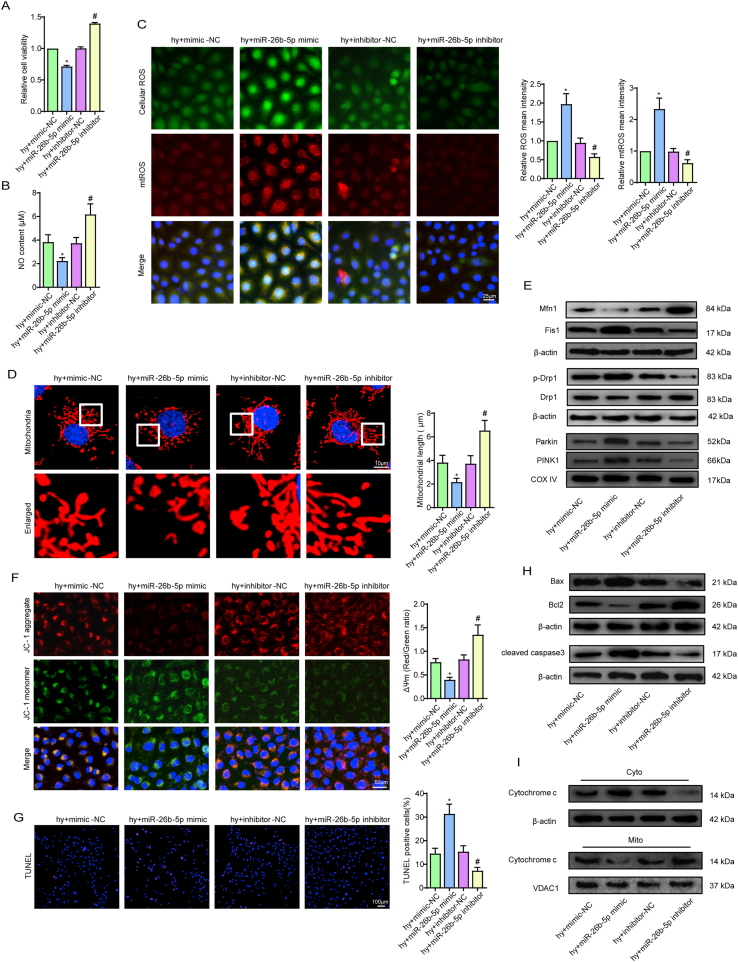
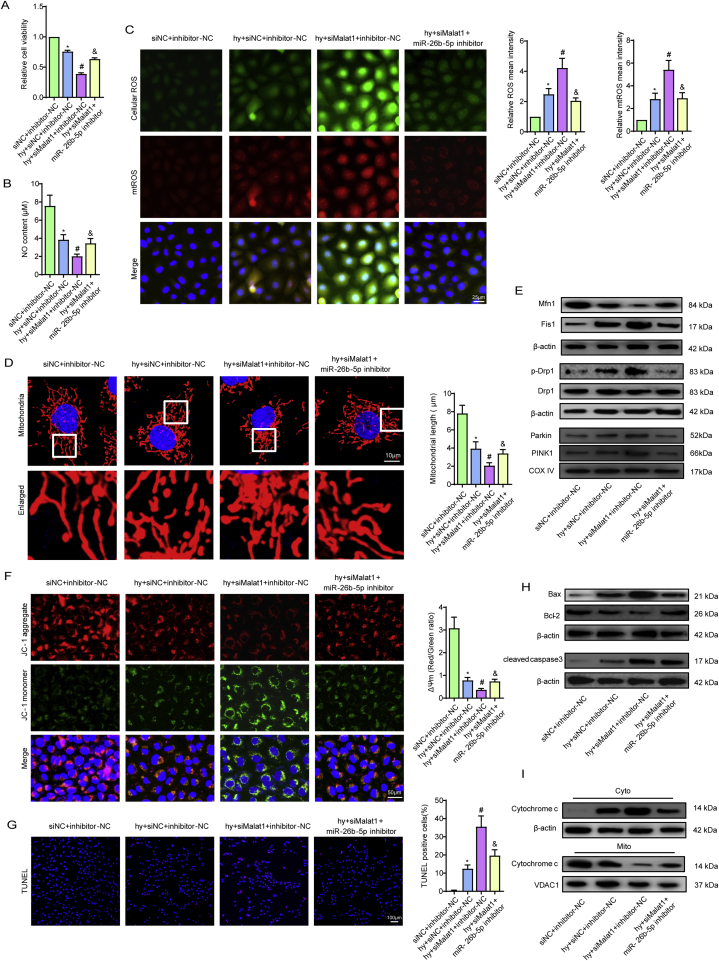
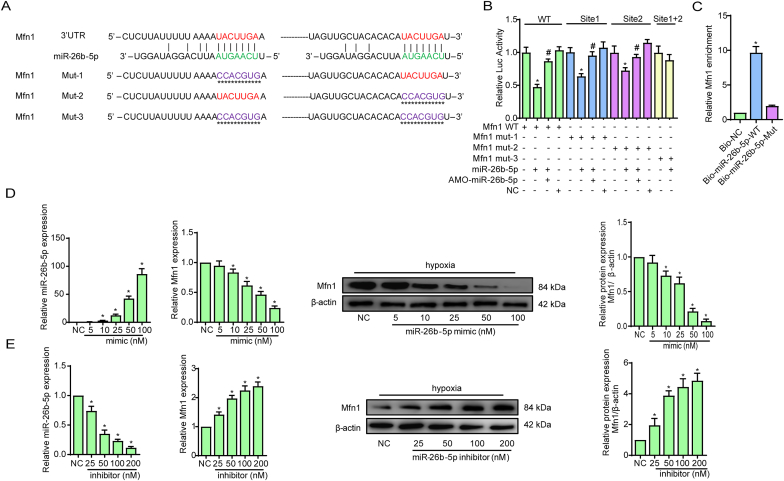
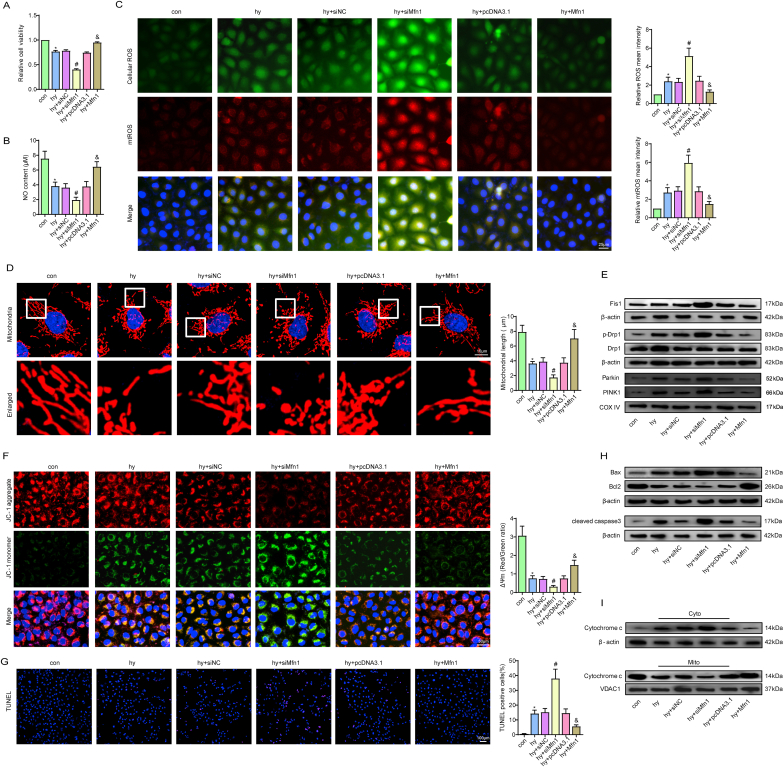
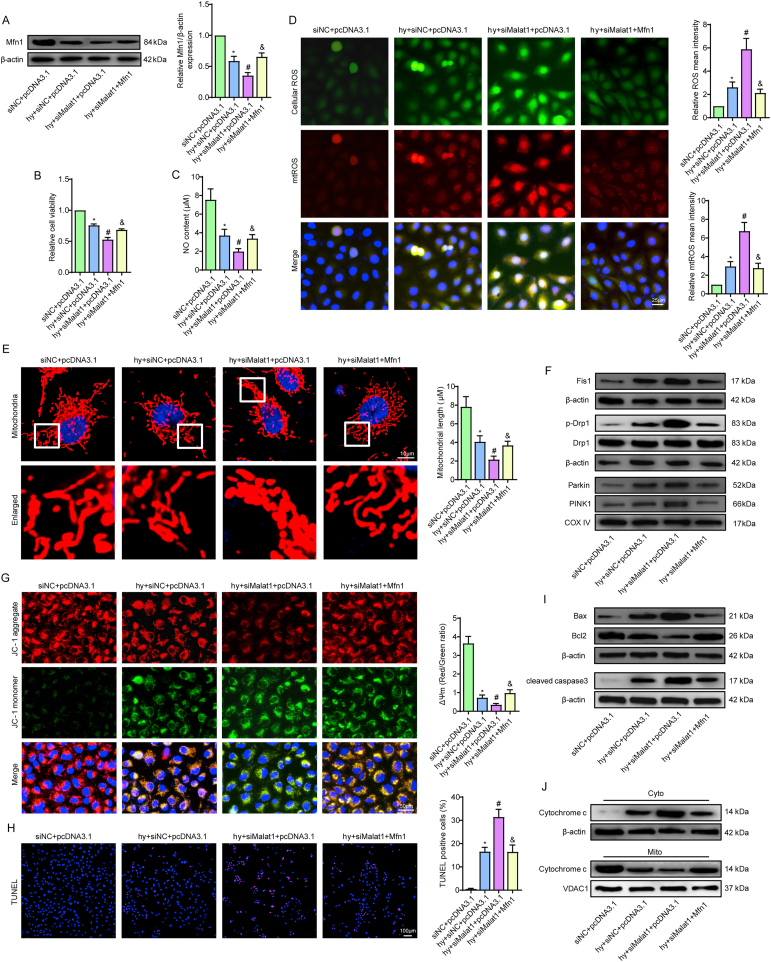
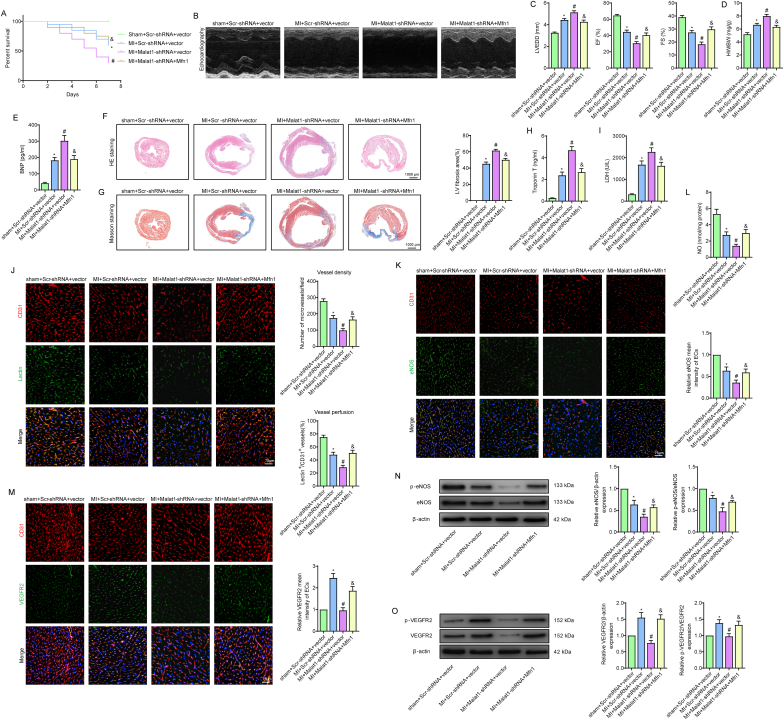
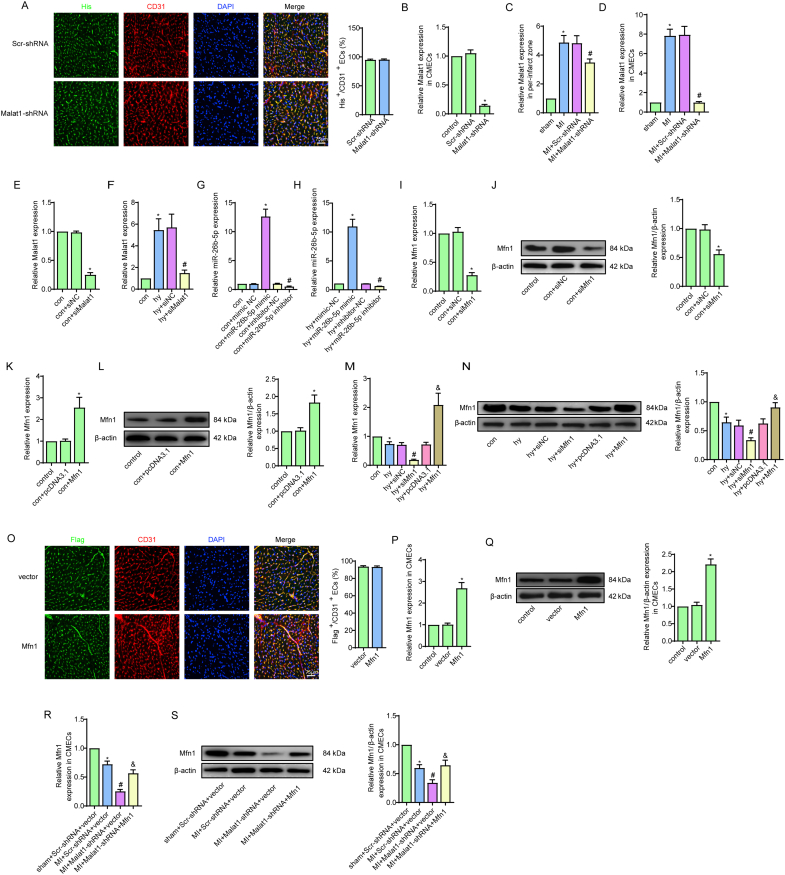
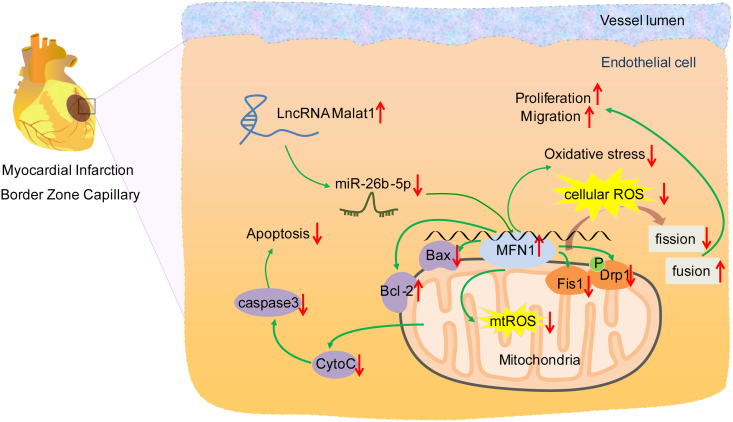
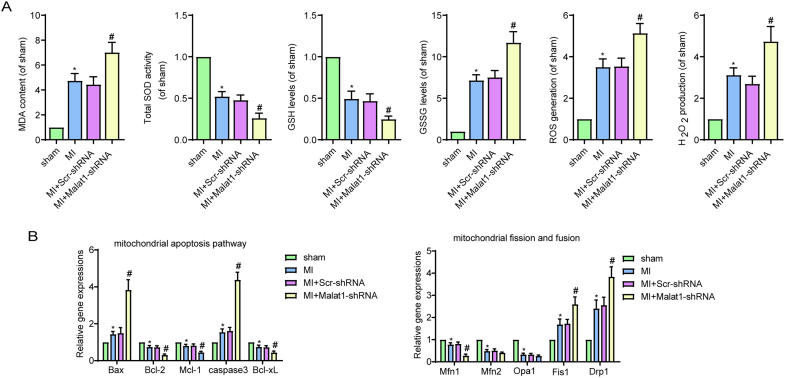
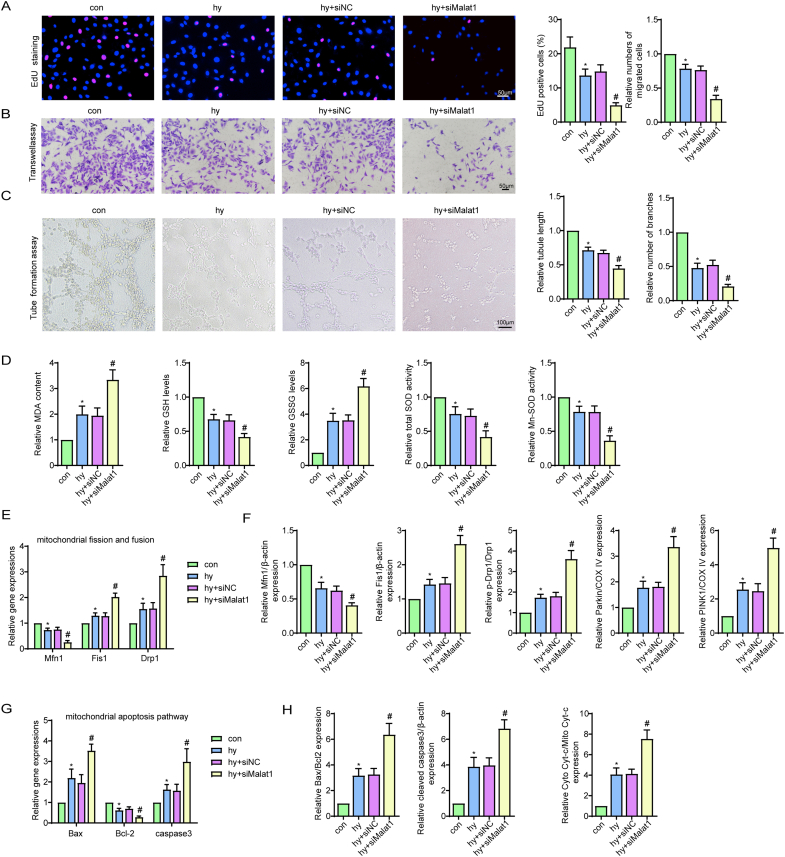
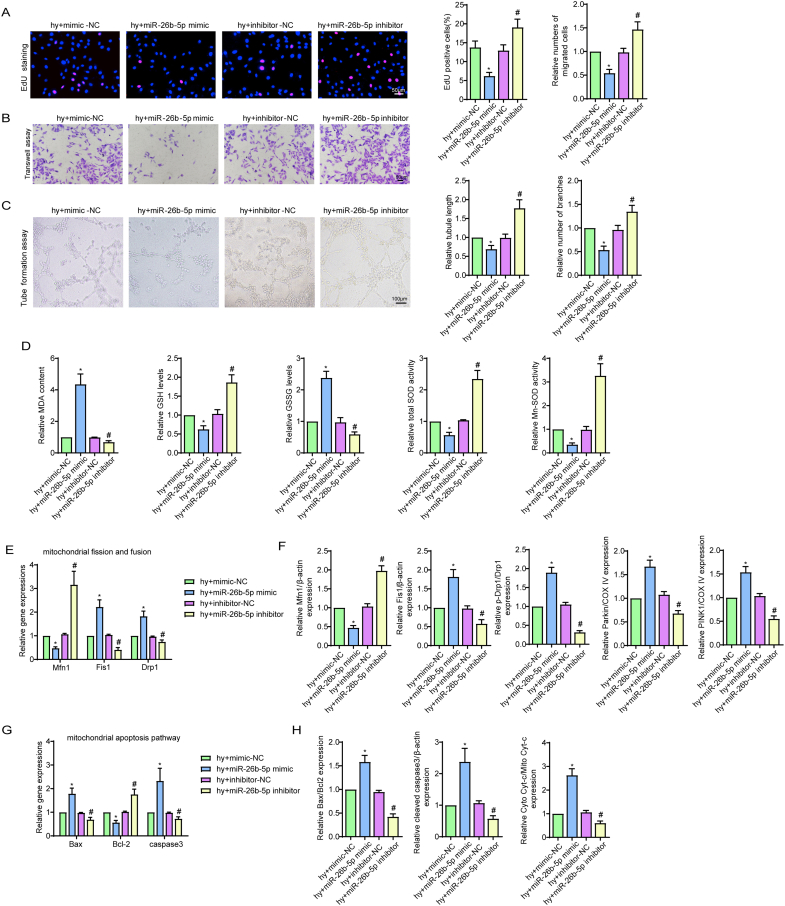
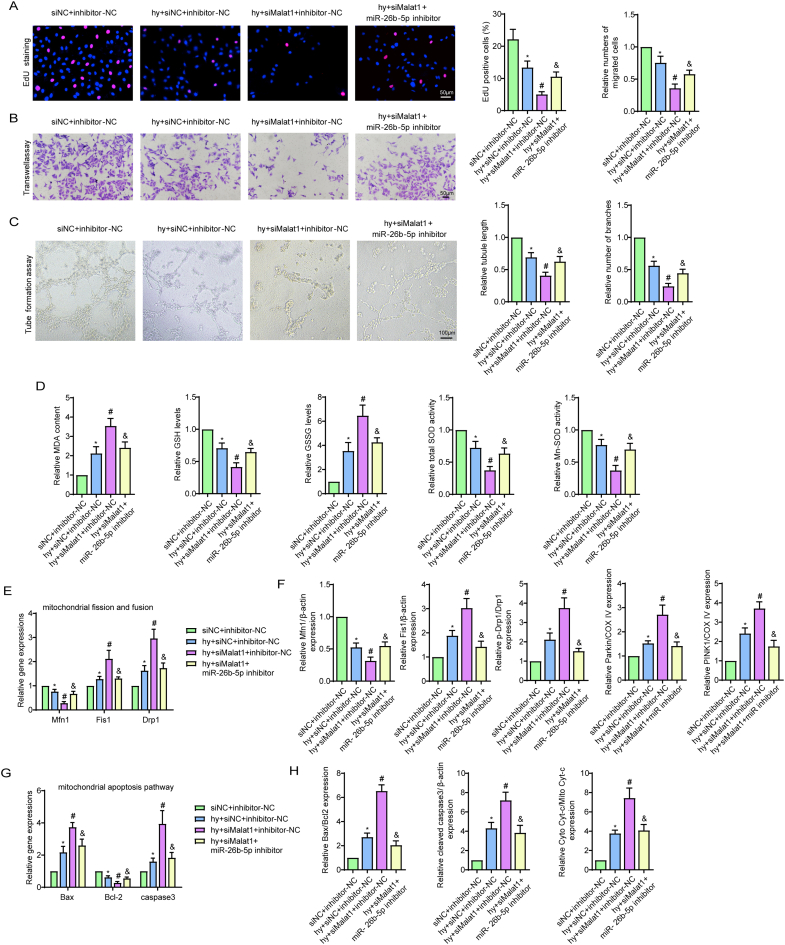
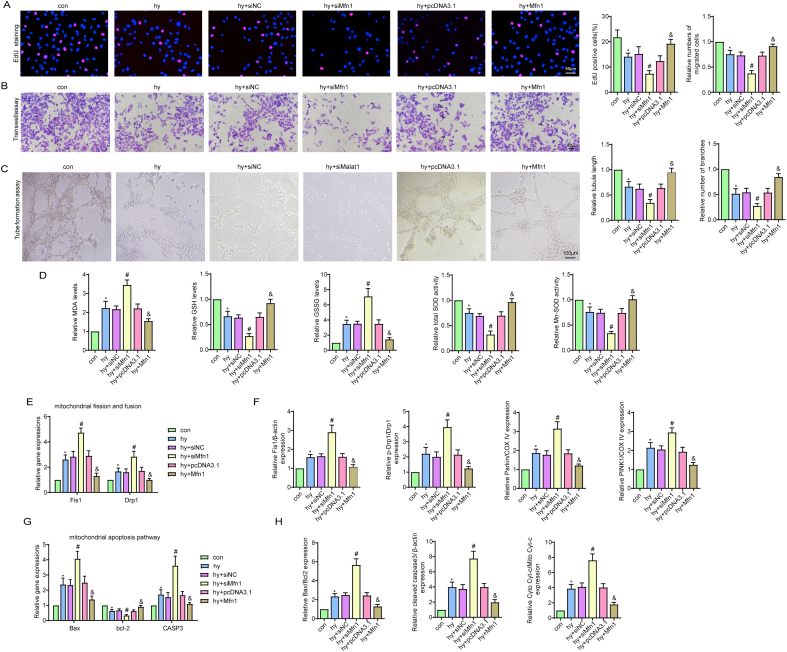
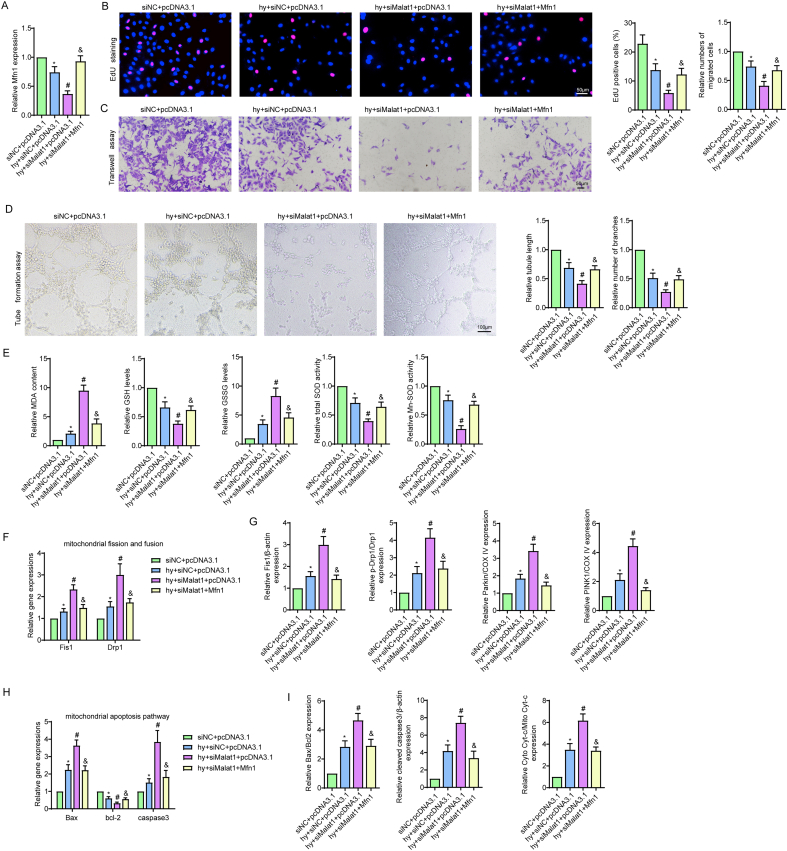
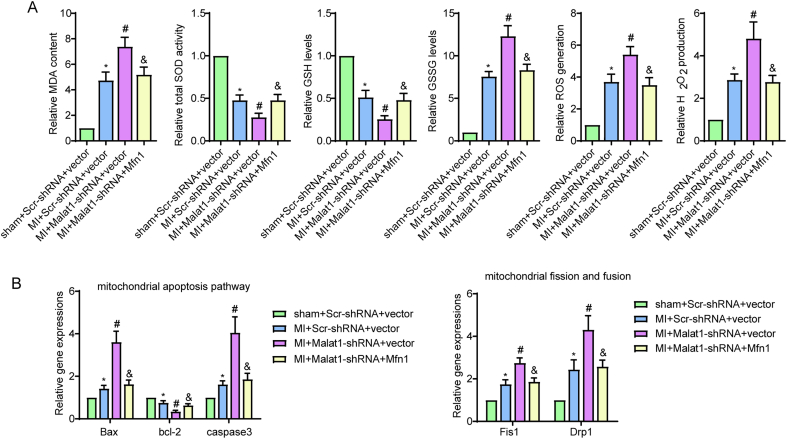
Methods and results: The present study found that Malat1 is upregulated in the border zone of infarction in mouse hearts, as well as in isolated cardiac microvascular endothelial cells (CMECs). Targeted knockdown of Malat1 in endothelial cells exacerbated oxidative stress, attenuated angiogenesis and microvascular perfusion, and as a result decreased cardiac function in MI mice. Further studies showed that silencing Malat1 obviously inhibited CMEC proliferation, migration and tube formation, which was at least in part attributed to disturbed mitochondrial dynamics and activation of the mitochondrial apoptosis pathway. Moreover, bioinformatic analyses, luciferase assays and pull-down assays indicated that Malat1 acted as a competing endogenous RNA (ceRNA) for miR-26b-5p and formed a signalling axis with Mfn1 to regulate mitochondrial dynamics and endothelial functions. Overexpression of Mfn1 markedly reversed the microvascular dysfunction and CMEC injuries that were aggravated by silencing Malat1 via inhibition of excessive mitochondrial fragments and mitochondria-dependent apoptosis.
Conclusions: The present study elucidated the functions and mechanisms of Malat1 in cardiac microcirculation repair after MI. The underlying mechanisms of the effects of Malat1 could be attributed to its blocking effects on miR-26b-5p/Mfn1 pathway-mediated mitochondrial dynamics and apoptosis.
Keywords: Cardiac microvascular dysfunction; Malat1; Mfn1; Myocardial infarction; miR-26b-5p.
Copyright © 2021 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- van Duijvenboden K. Conserved NPPB+ border zone switches from MEF2- to AP-1-driven gene program. Circulation. 2019;140(10):864–879. - PubMed
-
- Jayasuriya R. Role of Nrf 2 in MALAT1/HIF-1α loop on the regulation of angiogenesis in diabetic foot ulcer. Free Radic. Biol. Med. 2020;156:168–175. - PubMed
-
- Wang Y. Knockdown of MALAT1 attenuates high-glucose-induced angiogenesis and inflammation via endoplasmic reticulum stress in human retinal vascular endothelial cells. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2020;124:109699. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
