

New Insights on the Genetic Basis Underlying SHILCA Syndrome: Characterization of the NMNAT1 Pathological Alterations Due to Compound Heterozygous Mutations and Identification of a Novel Alternative Isoform
- PMID: 33668384
- PMCID: PMC7956282
- DOI: 10.3390/ijms22052262
New Insights on the Genetic Basis Underlying SHILCA Syndrome: Characterization of the NMNAT1 Pathological Alterations Due to Compound Heterozygous Mutations and Identification of a Novel Alternative Isoform
Abstract
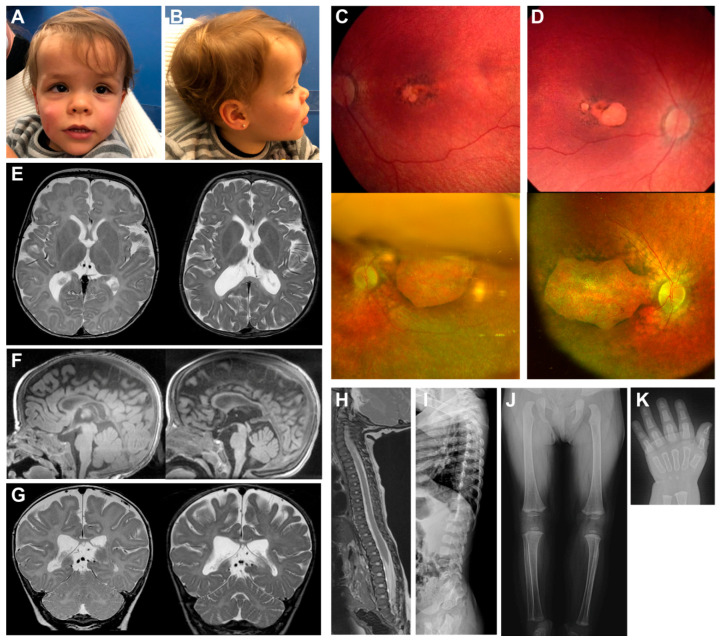
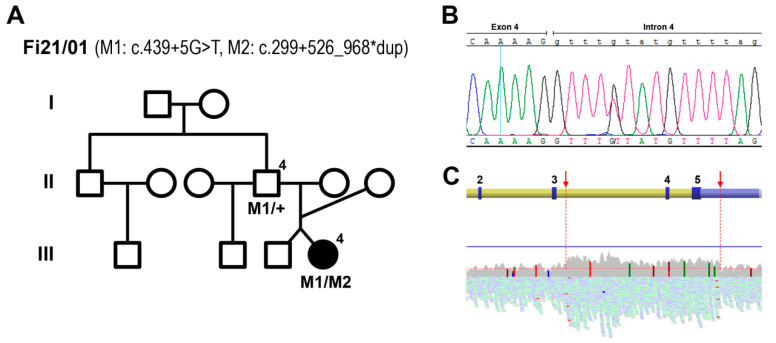
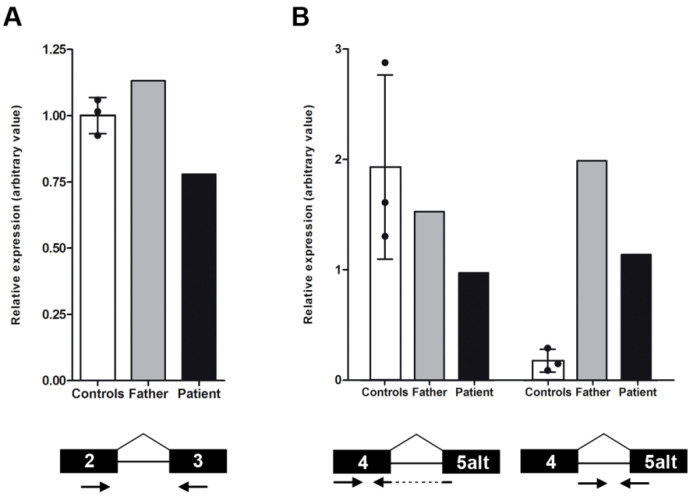
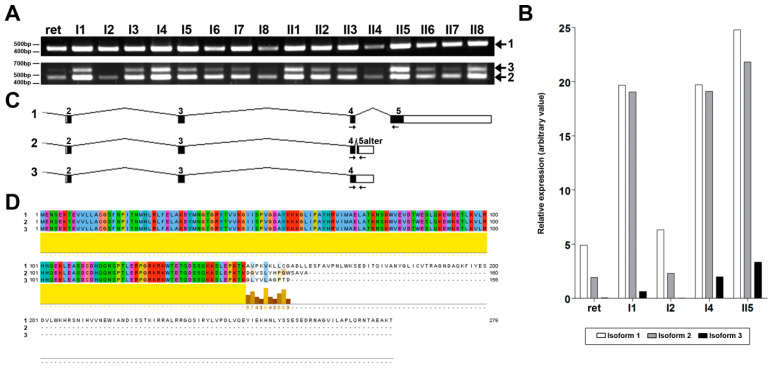
This study aims to genetically characterize a two-year-old patient suffering from multiple systemic abnormalities, including skeletal, nervous and developmental involvements and Leber congenital amaurosis (LCA). Genetic screening by next-generation sequencing identified two heterozygous pathogenic variants in nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1) as the molecular cause of the disease: c.439+5G>T and c.299+526_*968dup.This splice variant has never been reported to date, whereas pathogenic duplication has recently been associated with cases displaying an autosomal recessive disorder that includes a severe form of spondylo-epiphyseal dysplasia, sensorineural hearing loss, intellectual disability and LCA (SHILCA), as well as some brain anomalies. Our patient presented clinical manifestations which correlated strongly with this reported syndrome. To further study the possible transcriptional alterations resulting from these mutations, mRNA expression assays were performed in the patient and her father. The obtained results detected aberrant alternative transcripts and unbalanced levels of expression, consistent with severe systemic involvement. Moreover, these analyses also detected a novel NMNAT1 isoform, which is variably expressed in healthy human tissues. Altogether, these findings represent new evidence of the correlation of NMNAT1 and SHILCA syndrome, and provide additional insights into the healthy and pathogenic expression of this gene.
Keywords: Leber congenital amaurosis (LCA); developmental delay; hypomyelination; inherited retinal diseases; macular coloboma; nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1); sensorineural hearing loss; spondylo-epiphyseal dysplasia; spondylo-epiphyseal dysplasia, sensorineural hearing loss, intellectual disability and Leber congenital amaurosis (SHILCA); transcriptional alteration.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
An Alu-mediated duplication in NMNAT1, involved in NAD biosynthesis, causes a novel syndrome, SHILCA, affecting multiple tissues and organs.Hum Mol Genet. 2020 Aug 3;29(13):2250-2260. doi: 10.1093/hmg/ddaa112. Hum Mol Genet. 2020. PMID: 32533184
-
Novel compound heterozygous NMNAT1 variants associated with Leber congenital amaurosis.Mol Vis. 2014 Jun 2;20:753-9. eCollection 2014. Mol Vis. 2014. PMID: 24940029 Free PMC article.
-
NMNAT1 mutations cause Leber congenital amaurosis.Nat Genet. 2012 Sep;44(9):1040-5. doi: 10.1038/ng.2361. Epub 2012 Jul 29. Nat Genet. 2012. PMID: 22842227 Free PMC article.
-
Clinical and genetic findings in a family with NMNAT1-associated Leber congenital amaurosis: case report and review of the literature.Graefes Arch Clin Exp Ophthalmol. 2015 Dec;253(12):2239-46. doi: 10.1007/s00417-015-3174-0. Epub 2015 Oct 13. Graefes Arch Clin Exp Ophthalmol. 2015. PMID: 26464178 Review.
-
Clinical course of a Japanese girl with Leber congenital amaurosis associated with a novel nonsense pathogenic variant in NMNAT1: a case report and mini review.Ophthalmic Genet. 2022 Jun;43(3):400-408. doi: 10.1080/13816810.2021.2023195. Epub 2022 Jan 13. Ophthalmic Genet. 2022. PMID: 35026968 Review.
Cited by
-
Reduced nuclear NAD+ drives DNA damage and subsequent immune activation in the retina.Hum Mol Genet. 2022 May 4;31(9):1370-1388. doi: 10.1093/hmg/ddab324. Hum Mol Genet. 2022. PMID: 34750622 Free PMC article.
-
Role of Nuclear NAD+ in Retinal Homeostasis.Adv Exp Med Biol. 2023;1415:235-239. doi: 10.1007/978-3-031-27681-1_34. Adv Exp Med Biol. 2023. PMID: 37440039 Review.
-
Expression of NMNAT1 in the photoreceptors is sufficient to prevent NMNAT1-associated retinal degeneration.Mol Ther Methods Clin Dev. 2023 Apr 18;29:319-328. doi: 10.1016/j.omtm.2023.04.003. eCollection 2023 Jun 8. Mol Ther Methods Clin Dev. 2023. PMID: 37214313 Free PMC article.
-
Inherited Retinal Diseases.Int J Mol Sci. 2022 Nov 3;23(21):13467. doi: 10.3390/ijms232113467. Int J Mol Sci. 2022. PMID: 36362249 Free PMC article.
References
-
- Tsang S.H., Sharma T. Advances in Experimental Medicine and Biology. Springer International Publishing; Berlin/Heidelberg, Germany: 2018. Leber Congenital Amaurosis. - PubMed
-
- Koenekoop R.K., Lopez I., Allikmets R., Cremers F.P., Hollander A.I.D. Genetics, phenotypes, mechanisms and treatments for Leber congenital amaurosis: A paradigm shift. Expert Rev. Ophthalmol. 2008;3:397–415. doi: 10.1586/17469899.3.4.397. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
