

Application of a Clinical Workflow May Lead to Increased Diagnostic Precision in Hereditary Spastic Paraplegias and Cerebellar Ataxias: A Single Center Experience
- PMID: 33669240
- PMCID: PMC7919782
- DOI: 10.3390/brainsci11020246
Application of a Clinical Workflow May Lead to Increased Diagnostic Precision in Hereditary Spastic Paraplegias and Cerebellar Ataxias: A Single Center Experience
Abstract
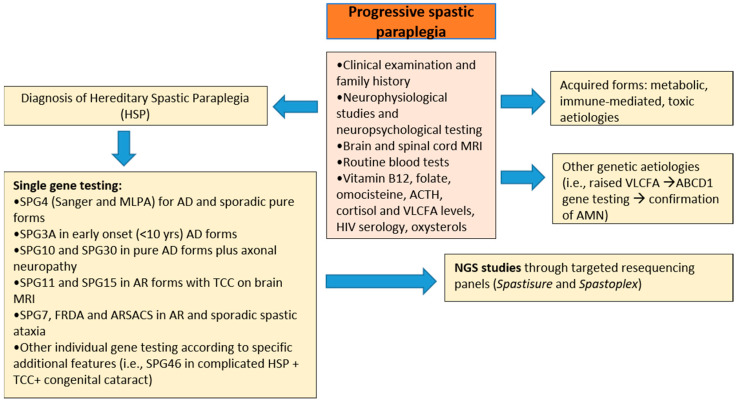
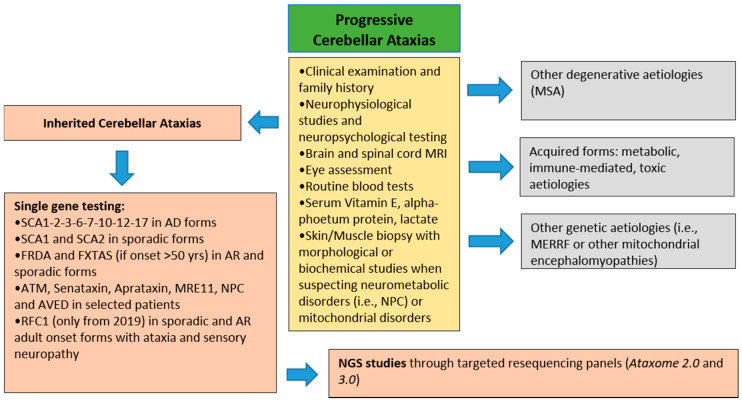
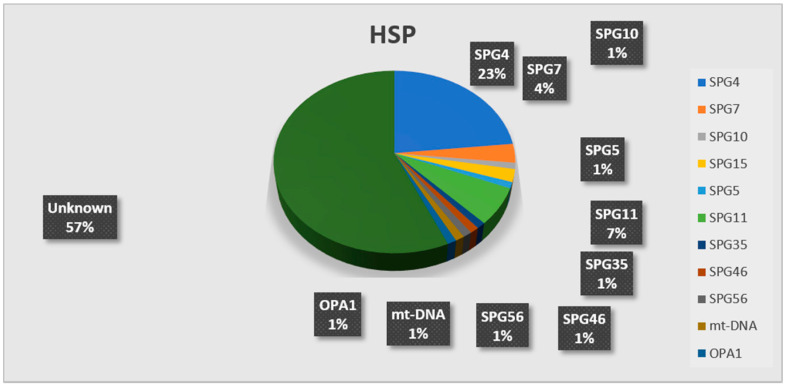
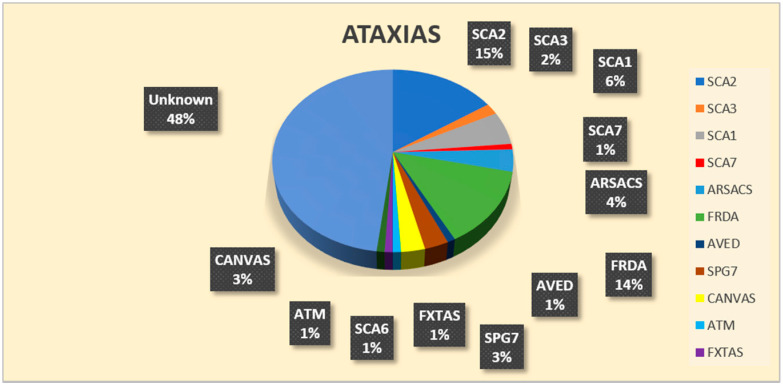
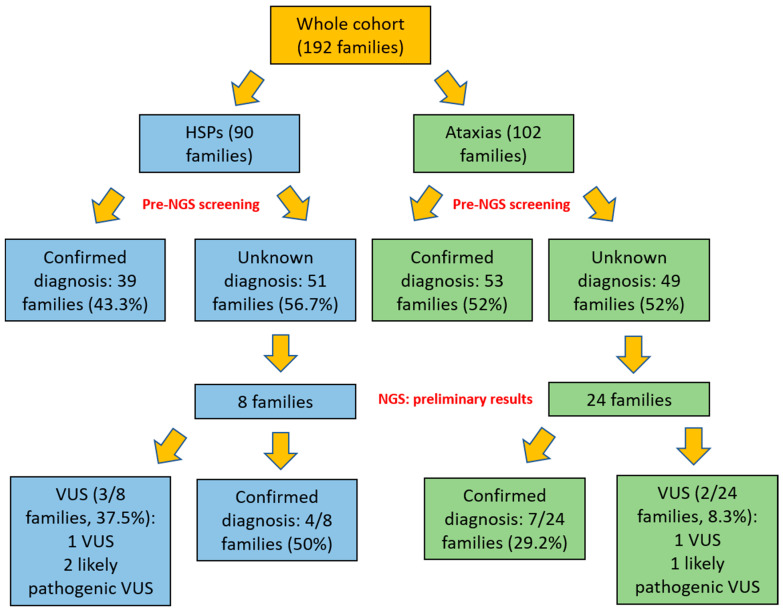
The molecular characterization of Hereditary Spastic Paraplegias (HSP) and inherited cerebellar ataxias (CA) is challenged by their clinical and molecular heterogeneity. The recent application of Next Generation Sequencing (NGS) technologies is increasing the diagnostic rate, which can be influenced by patients' selection. To assess if a clinical diagnosis of CA/HSP received in a third-level reference center might impact the molecular diagnostic yield, we retrospectively evaluated the molecular diagnostic rate reached in our center on 192 unrelated families (90 HSP and 102 CA) (i) before NGS and (ii) with the use of NGS gene panels. Overall, 46.3% of families received a genetic diagnosis by first-tier individual gene screening: 43.3% HSP and 50% spinocerebellar ataxias (SCA). The diagnostic rate was 56.7% in AD-HSP, 55.5% in AR-HSP, and 21.2% in sporadic HSP. On the other hand, 75% AD-, 52% AR- and 33% sporadic CA were diagnosed. So far, 32 patients (24 CA and 8 HSP) were further assessed by NGS gene panels, and 34.4% were diagnosed, including 29.2% CA and 50% HSP patients. Eleven novel gene variants classified as (likely) pathogenic were identified. Our results support the role of experienced clinicians in the diagnostic assessment and the clinical research of CA and HSP even in the next generation era.
Keywords: HSP; NGS; SCA; ataxia; hereditary spastic paraplegia; neurogenetics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Application of a custom NGS gene panel revealed a high diagnostic utility for molecular testing of hereditary ataxias.J Appl Genet. 2022 Sep;63(3):513-525. doi: 10.1007/s13353-022-00701-3. Epub 2022 May 19. J Appl Genet. 2022. PMID: 35588347
-
Next Generation Molecular Diagnosis of Hereditary Spastic Paraplegias: An Italian Cross-Sectional Study.Front Neurol. 2018 Dec 4;9:981. doi: 10.3389/fneur.2018.00981. eCollection 2018. Front Neurol. 2018. PMID: 30564185 Free PMC article.
-
[Ataxias and hereditary spastic paraplegias].Nervenarzt. 2017 Jul;88(7):720-727. doi: 10.1007/s00115-017-0357-4. Nervenarzt. 2017. PMID: 28600743 Review. German.
-
Genetic and Epidemiological Study of Adult Ataxia and Spastic Paraplegia in Eastern Quebec.Can J Neurol Sci. 2021 Sep;48(5):655-665. doi: 10.1017/cjn.2020.277. Epub 2021 Jan 5. Can J Neurol Sci. 2021. PMID: 33397523
-
Clinical application of next generation sequencing in hereditary spinocerebellar ataxia: increasing the diagnostic yield and broadening the ataxia-spasticity spectrum. A retrospective analysis.Neurogenetics. 2018 Jan;19(1):1-8. doi: 10.1007/s10048-017-0532-6. Epub 2017 Dec 6. Neurogenetics. 2018. PMID: 29209898 Review.
Cited by
-
NGS in Hereditary Ataxia: When Rare Becomes Frequent.Int J Mol Sci. 2021 Aug 6;22(16):8490. doi: 10.3390/ijms22168490. Int J Mol Sci. 2021. PMID: 34445196 Free PMC article.
-
Next-Generation Sequencing Technologies and Neurogenetic Diseases.Life (Basel). 2021 Apr 19;11(4):361. doi: 10.3390/life11040361. Life (Basel). 2021. PMID: 33921670 Free PMC article. Review.
-
Pluripotent Stem Cells as a Preclinical Cellular Model for Studying Hereditary Spastic Paraplegias.Int J Mol Sci. 2024 Feb 23;25(5):2615. doi: 10.3390/ijms25052615. Int J Mol Sci. 2024. PMID: 38473862 Free PMC article. Review.
-
Homozygous deep intronic variant in SNX14 cause autosomal recessive Spinocerebellar ataxia 20: a case report.Front Genet. 2023 Jul 6;14:1197681. doi: 10.3389/fgene.2023.1197681. eCollection 2023. Front Genet. 2023. PMID: 37485342 Free PMC article.
-
Clinical and genetic characterization of a Taiwanese cohort with spastic paraparesis combined with cerebellar involvement.Front Neurol. 2022 Sep 30;13:1005670. doi: 10.3389/fneur.2022.1005670. eCollection 2022. Front Neurol. 2022. PMID: 36247768 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
