

Primary Ciliary Dyskinesia Diagnostic Challenges: Understanding the Clinical Phenotype of the Puerto Rican RSPH4A Founder Mutation
- PMID: 33670432
- PMCID: PMC7918725
- DOI: 10.3390/diagnostics11020281
Primary Ciliary Dyskinesia Diagnostic Challenges: Understanding the Clinical Phenotype of the Puerto Rican RSPH4A Founder Mutation
Abstract
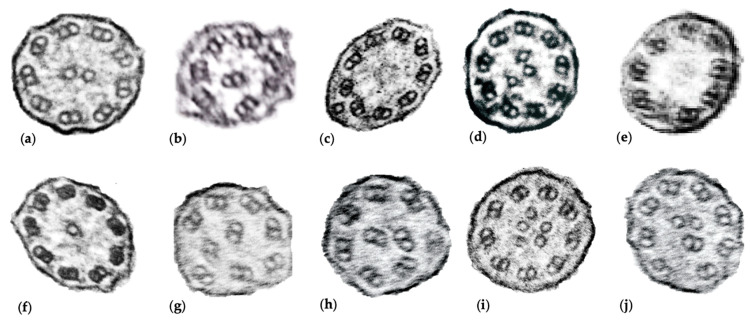
Primary ciliary dyskinesia (PCD) is a rare, heterogeneous ciliopathy resulting in chronic oto-sino-pulmonary disease, bronchiectasis, newborn respiratory distress, and laterality defects. PCD diagnosis can be achieved by following diagnostic algorithms that include electron microscopy, genetics, and ancillary testing. Genetic mutations in more than 45 genes, including RSPH4A, can lead to PCD. RSPH4A mutations located on chromosome six, affect radial spokes and results in central complex apparatus abnormalities. The RSPH4A [c.921 + 3_6delAAGT] founder mutation was described as one cause of PCD without laterality defects in Puerto Rico. Additionally, there are further diagnostic challenges present in the Puerto Rican population to diagnose PCD. We describe the demographics, clinical features, and RSPH4A genetic variants in 13 patients with clinical PCD affecting 11 Puerto Ricans from unrelated families.
Keywords: Puerto Rico; RSPH4A; cilia; founder mutation; primary ciliary dyskinesia.
Conflict of interest statement
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures

Similar articles
-
The Genetics of Primary Ciliary Dyskinesia in Puerto Rico.Diagnostics (Basel). 2022 May 2;12(5):1127. doi: 10.3390/diagnostics12051127. Diagnostics (Basel). 2022. PMID: 35626283 Free PMC article.
-
Primary Ciliary Dyskinesia: Ancestral Haplotypes Analysis of the RSPH4A Founder Mutation in Puerto Rico.Cureus. 2021 Sep 3;13(9):e17673. doi: 10.7759/cureus.17673. eCollection 2021 Sep. Cureus. 2021. PMID: 34513534 Free PMC article.
-
The RSPH4A Gene in Primary Ciliary Dyskinesia.Int J Mol Sci. 2023 Jan 18;24(3):1936. doi: 10.3390/ijms24031936. Int J Mol Sci. 2023. PMID: 36768259 Free PMC article. Review.
-
Founder mutation in RSPH4A identified in patients of Hispanic descent with primary ciliary dyskinesia.Hum Mutat. 2013 Oct;34(10):1352-6. doi: 10.1002/humu.22371. Epub 2013 Aug 6. Hum Mutat. 2013. PMID: 23798057 Free PMC article.
-
Value of transmission electron microscopy for primary ciliary dyskinesia diagnosis in the era of molecular medicine: Genetic defects with normal and non-diagnostic ciliary ultrastructure.Ultrastruct Pathol. 2017 Nov-Dec;41(6):373-385. doi: 10.1080/01913123.2017.1362088. Epub 2017 Sep 15. Ultrastruct Pathol. 2017. PMID: 28915070 Free PMC article. Review.
Cited by
-
The impact of primary ciliary dyskinesia on female and male fertility: a narrative review.Hum Reprod Update. 2023 May 2;29(3):347-367. doi: 10.1093/humupd/dmad003. Hum Reprod Update. 2023. PMID: 36721921 Free PMC article. Review.
-
Bronchiolitis Obliterans and Primary Ciliary Dyskinesia: What Is the Link?Cureus. 2021 Jun 11;13(6):e15591. doi: 10.7759/cureus.15591. eCollection 2021 Jun. Cureus. 2021. PMID: 34277212 Free PMC article.
-
The Genetics of Primary Ciliary Dyskinesia in Puerto Rico.Diagnostics (Basel). 2022 May 2;12(5):1127. doi: 10.3390/diagnostics12051127. Diagnostics (Basel). 2022. PMID: 35626283 Free PMC article.
-
Novel RSPH4A Variants Associated With Primary Ciliary Dyskinesia-Related Infertility in Three Chinese Families.Front Genet. 2022 Jun 22;13:922287. doi: 10.3389/fgene.2022.922287. eCollection 2022. Front Genet. 2022. PMID: 35812741 Free PMC article.
-
Advancing Primary Ciliary Dyskinesia Diagnosis through High-Speed Video Microscopy Analysis.Cells. 2024 Mar 24;13(7):567. doi: 10.3390/cells13070567. Cells. 2024. PMID: 38607006 Free PMC article.
References
-
- Zariwala M.A., Knowles M.R., Leigh M.W. Primary Ciliary Dyskinesia. GeneReviews; Seattle, WA, USA: 1993.
-
- Shapiro A.J., Davis S.D., Polineni D., Manion M., Rosenfeld M., Dell S.D., Chilvers M.A., Ferkol T.W., Zariwala M.A., Sagel S.D., et al. Diagnosis of Primary Ciliary Dyskinesia. An Official American Thoracic Society Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018;197:e24–e39. doi: 10.1164/rccm.201805-0819ST. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
