

APC Splicing Mutations Leading to In-Frame Exon 12 or Exon 13 Skipping Are Rare Events in FAP Pathogenesis and Define the Clinical Outcome
- PMID: 33670833
- PMCID: PMC7997234
- DOI: 10.3390/genes12030353
APC Splicing Mutations Leading to In-Frame Exon 12 or Exon 13 Skipping Are Rare Events in FAP Pathogenesis and Define the Clinical Outcome
Abstract
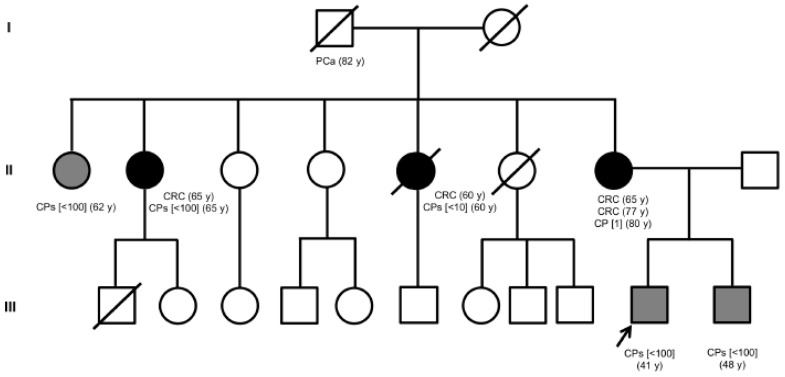
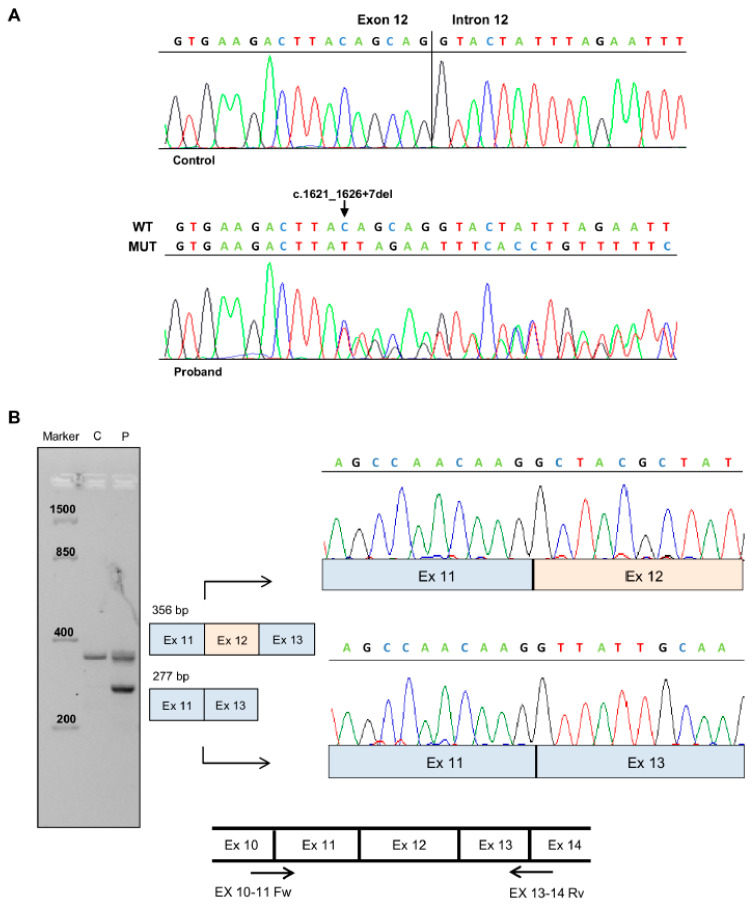
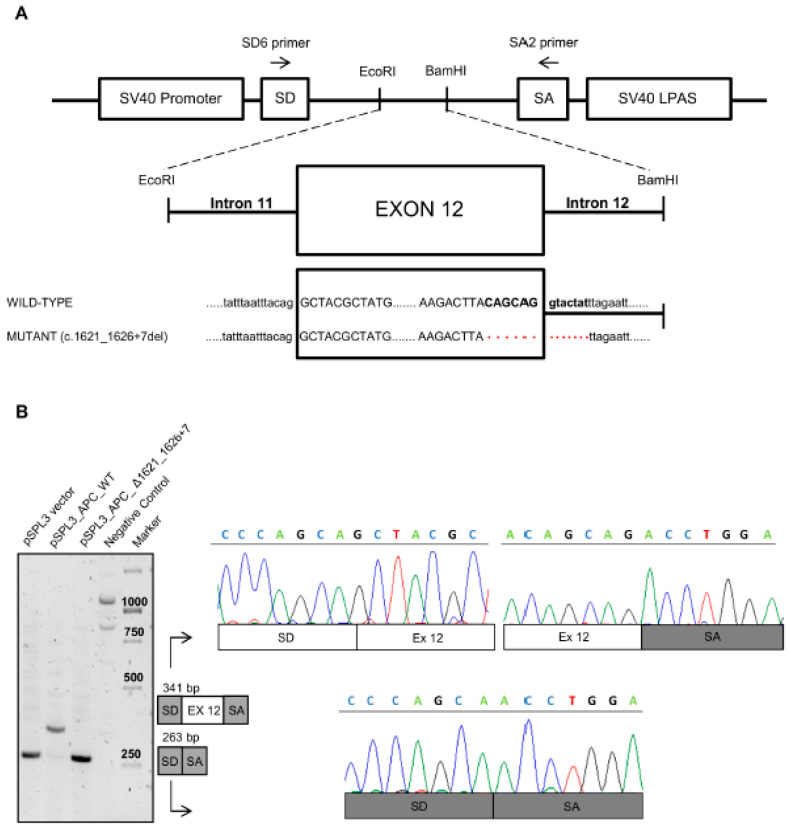
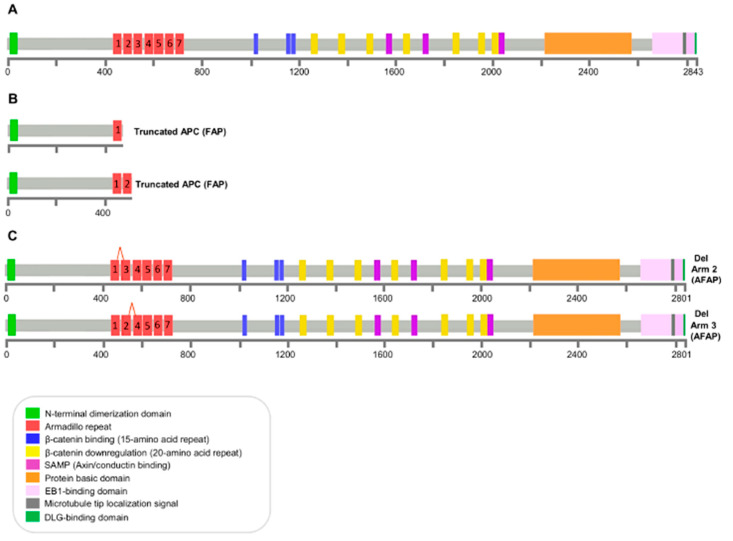
Familial adenomatous polyposis (FAP) is caused by germline mutations in the tumor suppressor gene APC. To date, nearly 2000 APC mutations have been described in FAP, most of which are predicted to result in truncated protein products. Mutations leading to aberrant APC splicing have rarely been reported. Here, we characterized a novel germline heterozygous splice donor site mutation in APC exon 12 (NM_000038.5: c.1621_1626+7del) leading to exon 12 skipping in an Italian family with the attenuated FAP (AFAP) phenotype. Moreover, we performed a literature meta-analysis of APC splicing mutations. We found that 119 unique APC splicing mutations, including the one described here, have been reported in FAP patients, 69 of which have been characterized at the mRNA level. Among these, only a small proportion (9/69) results in an in-frame protein, with four mutations causing skipping of exon 12 or 13 with loss of armadillo repeat 2 (ARM2) and 3 (ARM3), and five mutations leading to skipping of exon 5, 7, 8, or (partially) 9 with loss of regions not encompassing known functional domains. The APC splicing mutations causing skipping of exon 12 or 13 considered in this study cluster with the AFAP phenotype and reveal a potential molecular mechanism of pathogenesis in FAP disease.
Keywords: APC; FAP pathogenesis; exon skipping; familial adenomatous polyposis; splicing.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
A distinct APC pathogenic germline variant identified in a southern Thai family with familial adenomatous polyposis.BMC Med Genomics. 2021 Mar 19;14(1):87. doi: 10.1186/s12920-021-00933-y. BMC Med Genomics. 2021. PMID: 33740971 Free PMC article.
-
Synonymous mutation adenomatous polyposis coliΔ486s affects exon splicing and may predispose patients to adenomatous polyposis coli/mutY DNA glycosylase mutation‑negative familial adenomatous polyposis.Mol Med Rep. 2018 Dec;18(6):4931-4939. doi: 10.3892/mmr.2018.9495. Epub 2018 Sep 20. Mol Med Rep. 2018. PMID: 30272267 Free PMC article.
-
Intron 4 mutation in APC gene results in splice defect and attenuated FAP phenotype.Fam Cancer. 2004;3(1):35-40. doi: 10.1023/B:FAME.0000026824.85766.22. Fam Cancer. 2004. PMID: 15131404
-
Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): a review of the literature.Crit Rev Oncol Hematol. 2007 Feb;61(2):153-61. doi: 10.1016/j.critrevonc.2006.07.004. Epub 2006 Oct 24. Crit Rev Oncol Hematol. 2007. PMID: 17064931 Review.
-
The genetics of familial adenomatous polyposis (FAP) and MutYH-associated polyposis (MAP).Acta Gastroenterol Belg. 2011 Sep;74(3):421-6. Acta Gastroenterol Belg. 2011. PMID: 22103048 Review.
Cited by
-
De novo familial adenomatous polyposis with germline double heterozygosity of APC/BRCA2: a case report and literature review.Hered Cancer Clin Pract. 2025 Feb 21;23(1):6. doi: 10.1186/s13053-025-00306-x. Hered Cancer Clin Pract. 2025. PMID: 39985003 Free PMC article.
-
Clinical Assessment and Genetic Testing for Hereditary Polyposis Syndromes in an Italian Cohort of Patients with Colorectal Polyps.Cancers (Basel). 2024 Oct 26;16(21):3617. doi: 10.3390/cancers16213617. Cancers (Basel). 2024. PMID: 39518056 Free PMC article.
-
The implications of alternative pre-mRNA splicing in cell signal transduction.Exp Mol Med. 2023 Apr;55(4):755-766. doi: 10.1038/s12276-023-00981-7. Epub 2023 Apr 3. Exp Mol Med. 2023. PMID: 37009804 Free PMC article. Review.
-
Regulation of Pre-mRNA Splicing: Indispensable Role of Post-Translational Modifications of Splicing Factors.Life (Basel). 2023 Feb 21;13(3):604. doi: 10.3390/life13030604. Life (Basel). 2023. PMID: 36983760 Free PMC article. Review.
References
-
- Church J. Molecular Genetics of Colorectal Cancer. Semin. Colon Rectal Surg. 2016;27:172–175. doi: 10.1053/j.scrs.2016.04.013. - DOI
-
- Caspari R., Olschwang S., Friedl W., Mandl M., Boisson C., Böker T., Augustin A., Kadmon M., Möslein G., Thomas G. Familial Adenomatous Polyposis: Desmoid Tumours and Lack of Ophthalmic Lesions (CHRPE) Associated with APC Mutations beyond Codon 1444. Hum. Mol. Genet. 1995;4:337–340. doi: 10.1093/hmg/4.3.337. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
