

Innovative Therapeutic Approaches for Duchenne Muscular Dystrophy
- PMID: 33671409
- PMCID: PMC7922390
- DOI: 10.3390/jcm10040820
Innovative Therapeutic Approaches for Duchenne Muscular Dystrophy
Abstract
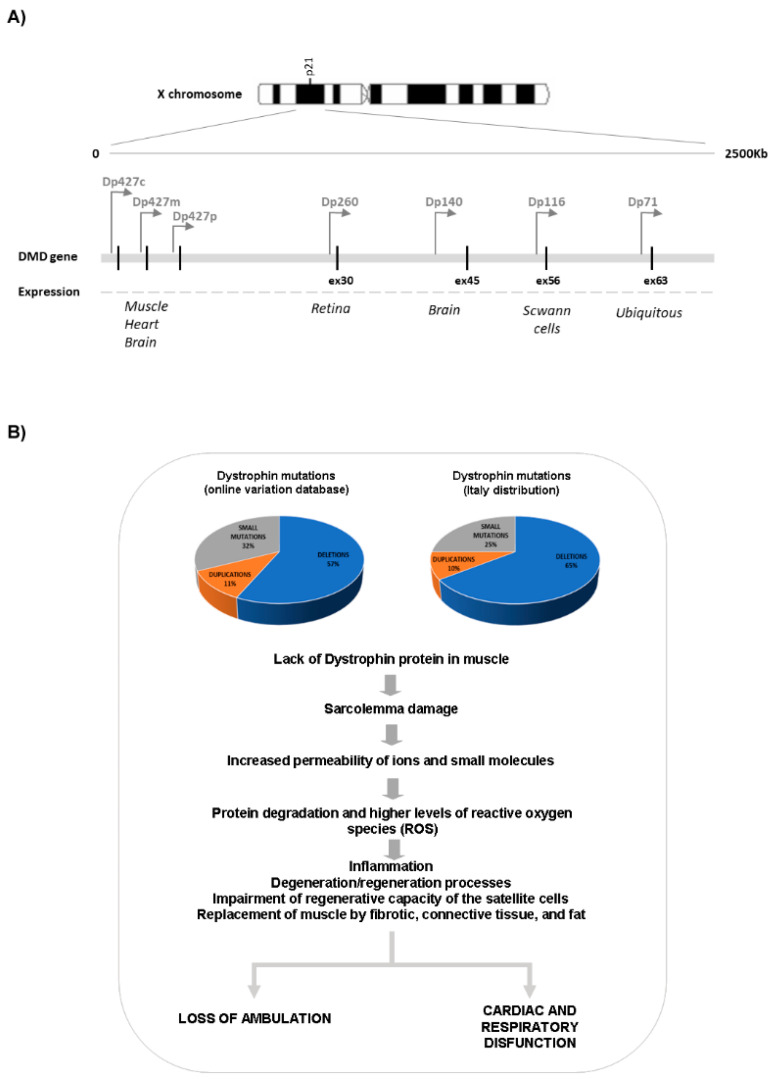
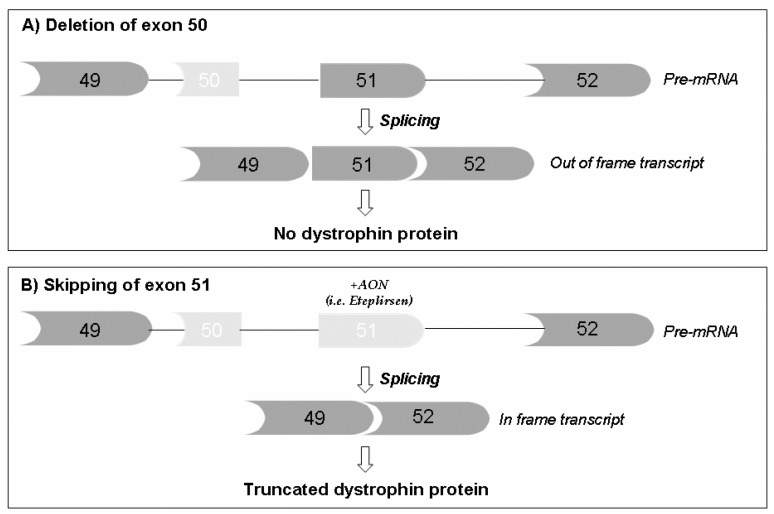
Duchenne muscular dystrophy (DMD) is the most common childhood muscular dystrophy affecting ~1:5000 live male births. Following the identification of pathogenic variations in the dystrophin gene in 1986, the underlining genotype/phenotype correlations emerged and the role of the dystrophin protein was elucidated in skeletal, smooth, and cardiac muscles, as well as in the brain. When the dystrophin protein is absent or quantitatively or qualitatively modified, the muscle cannot sustain the stress of repeated contractions. Dystrophin acts as a bridging and anchoring protein between the sarcomere and the sarcolemma, and its absence or reduction leads to severe muscle damage that eventually cannot be repaired, with its ultimate substitution by connective tissue and fat. The advances of an understanding of the molecular pathways affected in DMD have led to the development of many therapeutic strategies that tackle different aspects of disease etiopathogenesis, which have recently led to the first successful approved orphan drugs for this condition. The therapeutic advances in this field have progressed exponentially, with second-generation drugs now entering in clinical trials as gene therapy, potentially providing a further effective approach to the condition.
Keywords: Duchenne muscular dystrophy; antisense oligonucleotide chemistry; dystrophin restoration; exon-skipping; gene therapy; innovative clinical trials; stop codon reversion.
Conflict of interest statement
A.F. is Principal Investigator and F.F. is Study Coordinator of the Sarepta Therapeutics Essence and MIS51ON clinical trial for DMD. A.F. is the PI of ongoing grants on DMD diagnosis funded by PTC Therapeutics and Sarepta Therapeutics.
Figures
References
-
- Koenig M., Hoffman E.P., Bertelson C.J., Monaco A.P., Feener C., Kunkel L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–517. doi: 10.1016/0092-8674(87)90504-6. - DOI - PubMed
-
- Barseghyan H., Tang W., Wang R.T., Almalvez M., Segura E., Bramble M.S., Lipson A., Douine E.D., Lee H., Délot E.C., et al. Next-generation mapping: A novel approach for detection of pathogenic structural variants with a potential utility in clinical diagnosis. Genome Med. 2017;9:90. doi: 10.1186/s13073-017-0479-0. - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
