

Oxidative Stress in the Tumor Microenvironment and Its Relevance to Cancer Immunotherapy
- PMID: 33673398
- PMCID: PMC7956301
- DOI: 10.3390/cancers13050986
Oxidative Stress in the Tumor Microenvironment and Its Relevance to Cancer Immunotherapy
Abstract
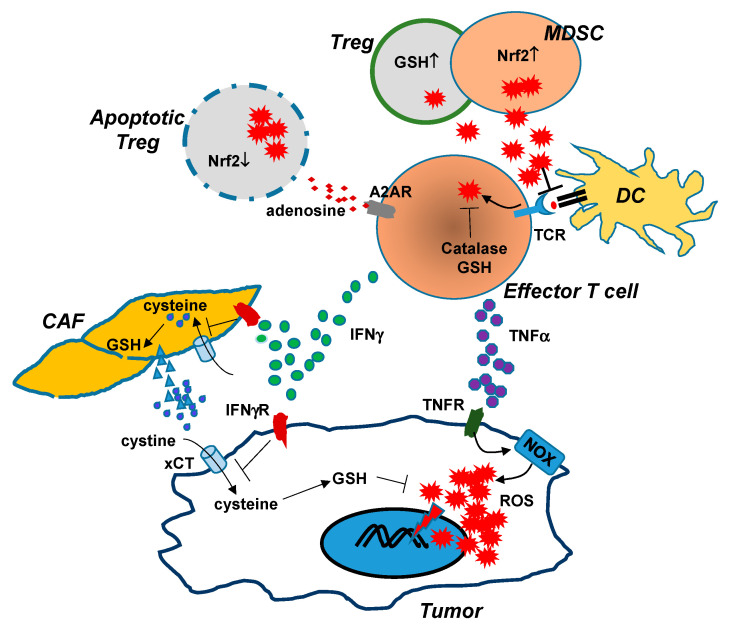
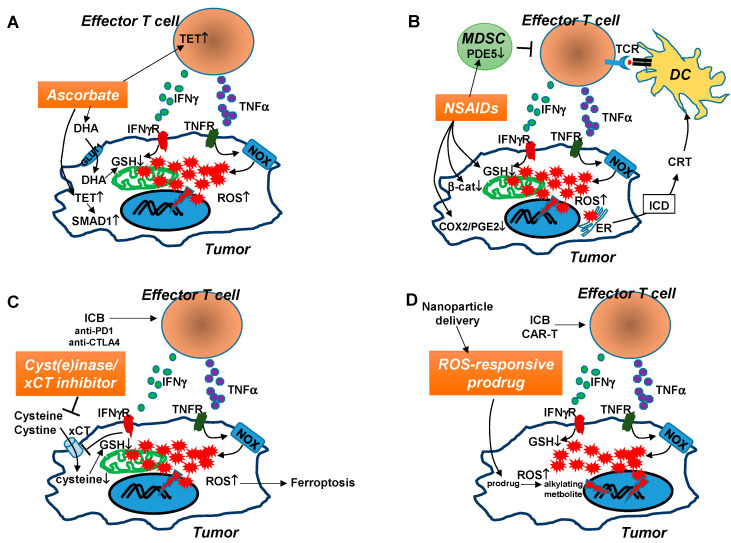
It has been well-established that cancer cells are under constant oxidative stress, as reflected by elevated basal level of reactive oxygen species (ROS), due to increased metabolism driven by aberrant cell growth. Cancer cells can adapt to maintain redox homeostasis through a variety of mechanisms. The prevalent perception about ROS is that they are one of the key drivers promoting tumor initiation, progression, metastasis, and drug resistance. Based on this notion, numerous antioxidants that aim to mitigate tumor oxidative stress have been tested for cancer prevention or treatment, although the effectiveness of this strategy has yet to be established. In recent years, it has been increasingly appreciated that ROS have a complex, multifaceted role in the tumor microenvironment (TME), and that tumor redox can be targeted to amplify oxidative stress inside the tumor to cause tumor destruction. Accumulating evidence indicates that cancer immunotherapies can alter tumor redox to intensify tumor oxidative stress, resulting in ROS-dependent tumor rejection. Herein we review the recent progresses regarding the impact of ROS on cancer cells and various immune cells in the TME, and discuss the emerging ROS-modulating strategies that can be used in combination with cancer immunotherapies to achieve enhanced antitumor effects.
Keywords: immunotherapy; oxidative stress; reactive oxygen species; tumor microenvironment.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Reczek C.R., Chandel N.S. The Two Faces of Reactive Oxygen Species in Cancer. Annu. Rev. Cancer Biol. 2017;1:79–98. doi: 10.1146/annurev-cancerbio-041916-065808. - DOI
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
