

The Dying Forward Hypothesis of ALS: Tracing Its History
- PMID: 33673524
- PMCID: PMC7997258
- DOI: 10.3390/brainsci11030300
The Dying Forward Hypothesis of ALS: Tracing Its History
Abstract
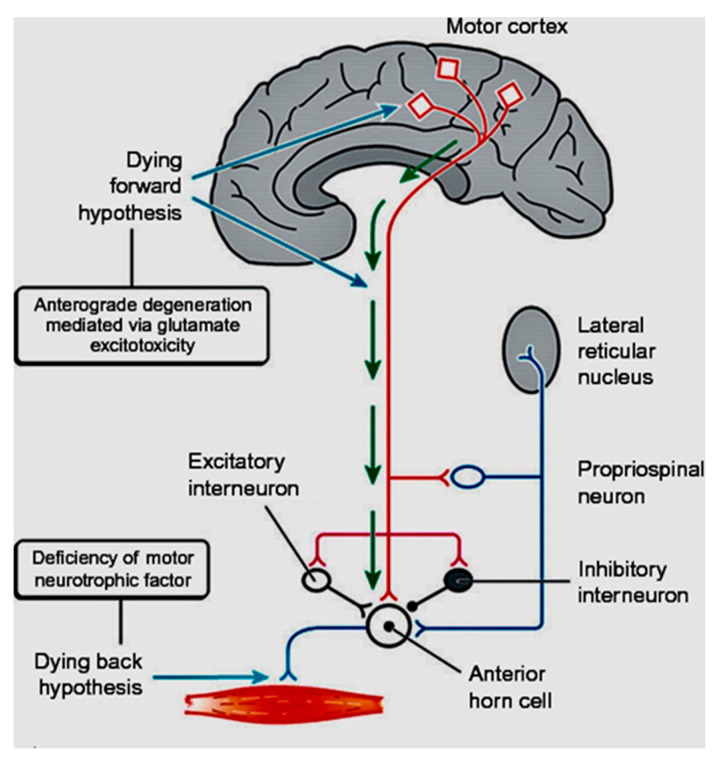
The site of origin of amyotrophic lateral sclerosis (ALS), although unsettled, is increasingly recognized as being cortico-fugal, which is a dying-forward process primarily starting in the corticomotoneuronal system. A variety of iterations of this concept date back to over 150 years. Recently, the hallmark TAR DNA-binding protein 43 (TDP-43) pathology, seen in >95% of patients with ALS, has been shown to be largely restricted to corticofugal projecting neurons ("dying forward"). Possibly, soluble but toxic cytoplasmic TDP-43 could enter the axoplasm of Betz cells, subsequently causing dysregulation of nuclear protein in the lower brainstem and spinal cord anterior horn cells. As the disease progresses, cortical involvement in ALS becomes widespread, including or starting with frontotemporal dementia, implying a broader view of ALS as a brain disease. The onset at the motor and premotor cortices should be considered a nidus at the edge of multiple cortical networks which eventually become disrupted, causing failure of a widespread cortical connectome.
Keywords: TDP-43; amyotrophic lateral sclerosis; dying-forward; frontotemporal dementia; neural networks; neurodegeneration.
Conflict of interest statement
The author declares no conflict of interest.
Figures
References
-
- Snowden J.S., Harris J., Richardson A., Rollinson S., Thompson J.C., Neary D., Mann D.M., Pickering-Brown S. Frontotemporal dementia with amyotrophic lateral sclerosis: A clinical comparison of patients with and without repeat expansions in C9orf72. Amyotroph. Lateral Scler. Front. Degener. 2013;14:172–176. doi: 10.3109/21678421.2013.765485. - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
