

Subversion of Niche-Signalling Pathways in Colorectal Cancer: What Makes and Breaks the Intestinal Stem Cell
- PMID: 33673710
- PMCID: PMC7957493
- DOI: 10.3390/cancers13051000
Subversion of Niche-Signalling Pathways in Colorectal Cancer: What Makes and Breaks the Intestinal Stem Cell
Abstract
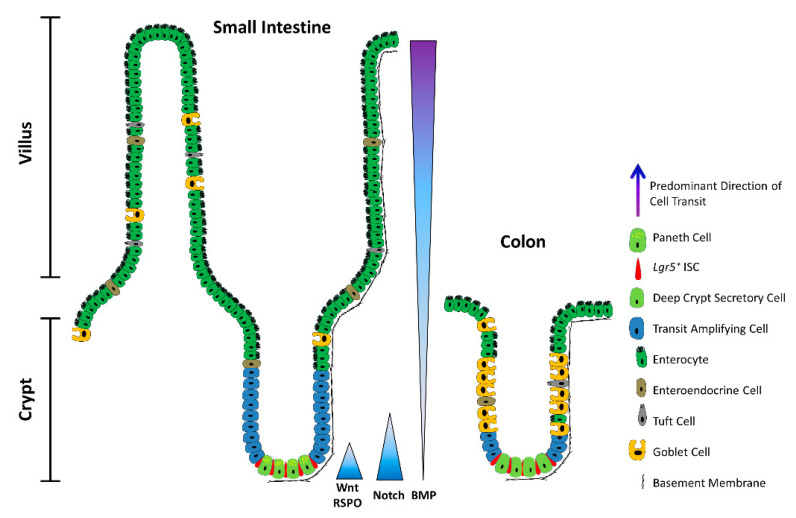
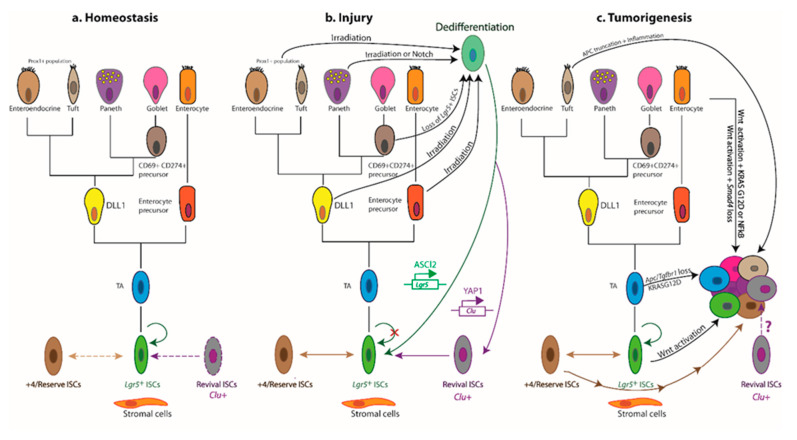
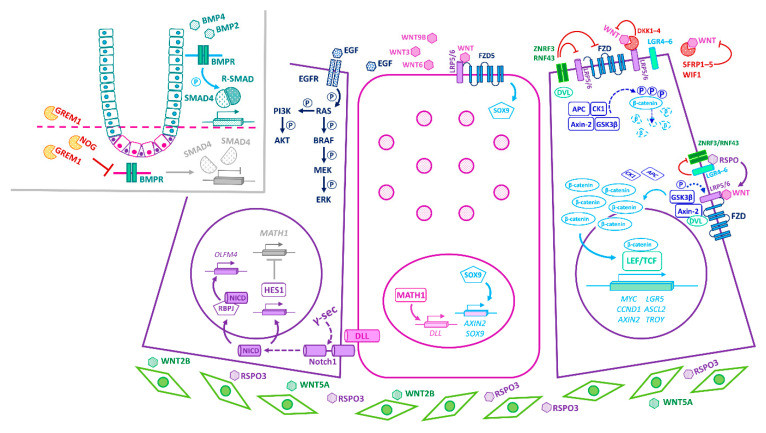
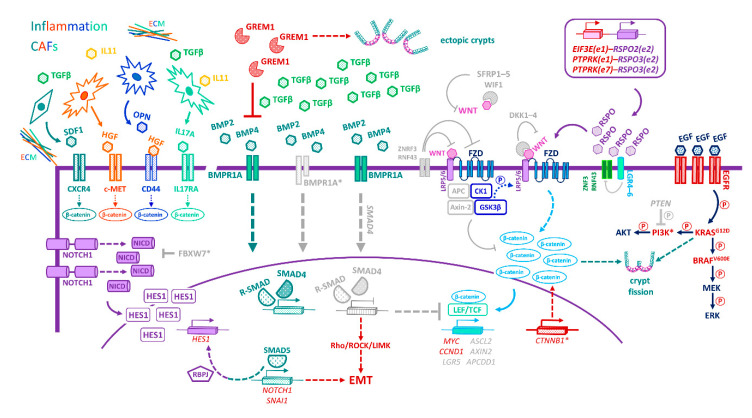
The intestinal epithelium fulfils pleiotropic functions in nutrient uptake, waste elimination, and immune surveillance while also forming a barrier against luminal toxins and gut-resident microbiota. Incessantly barraged by extraneous stresses, the intestine must continuously replenish its epithelial lining and regenerate the full gamut of specialized cell types that underpin its functions. Homeostatic remodelling is orchestrated by the intestinal stem cell (ISC) niche: a convergence of epithelial- and stromal-derived cues, which maintains ISCs in a multipotent state. Following demise of homeostatic ISCs post injury, plasticity is pervasive among multiple populations of reserve stem-like cells, lineage-committed progenitors, and/or fully differentiated cell types, all of which can contribute to regeneration and repair. Failure to restore the epithelial barrier risks seepage of toxic luminal contents, resulting in inflammation and likely predisposing to tumour formation. Here, we explore how homeostatic niche-signalling pathways are subverted in tumorigenesis, enabling ISCs to gain autonomy from niche restraints ("ISC emancipation") and transform into cancer stem cells capable of driving tumour initiation, progression, and therapy resistance. We further consider the implications of the pervasive plasticity of the intestinal epithelium for the trajectory of colorectal cancer, the emergence of distinct molecular subtypes, the propensity to metastasize, and the development of effective therapeutic strategies.
Keywords: BMP; Notch; Wnt; YAP; cancer stem cells (CSCs); colorectal cancer (CRC); consensus molecular subtypes (CMS); intestinal stem cell (ISC) niche; intestinal stem cells (ISCs); regeneration.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Sasaki N., Sachs N., Wiebrands K., Ellenbroek S.I., Fumagalli A., Lyubimova A., Begthel H., van den Born M., van Es J.H., Karthaus W.R., et al. Reg4+ deep crypt secretory cells function as epithelial niche for Lgr5+ stem cells in colon. Proc. Natl. Acad. Sci. USA. 2016;113:E5399–E5407. doi: 10.1073/pnas.1607327113. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
