

Single-cell transcriptomic analysis of mIHC images via antigen mapping
- PMID: 33674303
- PMCID: PMC7935366
- DOI: 10.1126/sciadv.abc5464
Single-cell transcriptomic analysis of mIHC images via antigen mapping
Abstract
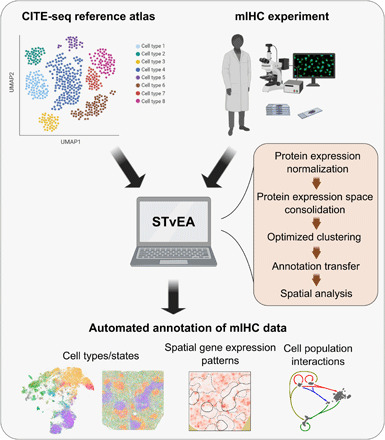
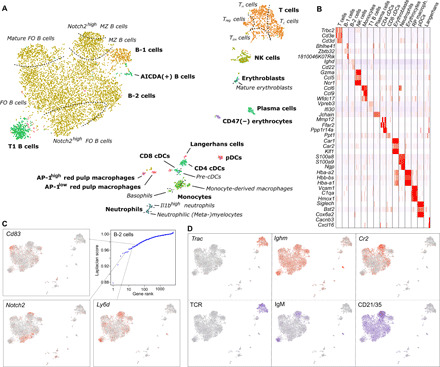
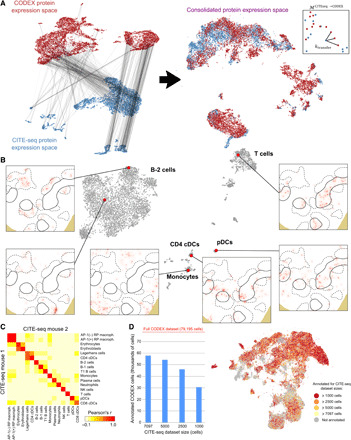
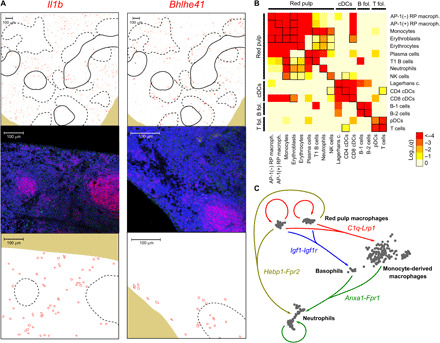
Highly multiplexed immunohistochemistry (mIHC) enables the staining and quantification of dozens of antigens in a tissue section with single-cell resolution. However, annotating cell populations that differ little in the profiled antigens or for which the antibody panel does not include specific markers is challenging. To overcome this obstacle, we have developed an approach for enriching mIHC images with single-cell RNA sequencing data, building upon recent experimental procedures for augmenting single-cell transcriptomes with concurrent antigen measurements. Spatially-resolved Transcriptomics via Epitope Anchoring (STvEA) performs transcriptome-guided annotation of highly multiplexed cytometry datasets. It increases the level of detail in histological analyses by enabling the systematic annotation of nuanced cell populations, spatial patterns of transcription, and interactions between cell types. We demonstrate the utility of STvEA by uncovering the architecture of poorly characterized cell types in the murine spleen using published cytometry and mIHC data of this organ.
Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC).
Figures

References
-
- Stack E. C., Wang C., Roman K. A., Hoyt C. C., Multiplexed immunohistochemistry, imaging, and quantitation: A review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods 70, 46–58 (2014). - PubMed
-
- Cornett D. S., Reyzer M. L., Chaurand P., Caprioli R. M., MALDI imaging mass spectrometry: Molecular snapshots of biochemical systems. Nat. Methods 4, 828–833 (2007). - PubMed
-
- Giesen C., Wang H. A. O., Schapiro D., Zivanovic N., Jacobs A., Hattendorf B., Schüffler P. J., Grolimund D., Buhmann J. M., Brandt S., Varga Z., Wild P. J., Günther D., Bodenmiller B., Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 11, 417–422 (2014). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
