

Preclinical Efficacy and Anti-Inflammatory Mechanisms of Action of the Bruton Tyrosine Kinase Inhibitor Rilzabrutinib for Immune-Mediated Disease
- PMID: 33674445
- PMCID: PMC7980532
- DOI: 10.4049/jimmunol.2001130
Preclinical Efficacy and Anti-Inflammatory Mechanisms of Action of the Bruton Tyrosine Kinase Inhibitor Rilzabrutinib for Immune-Mediated Disease
Abstract
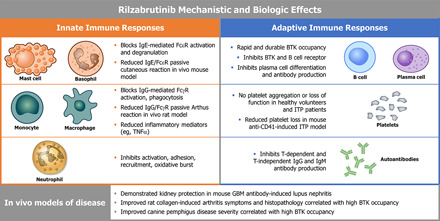
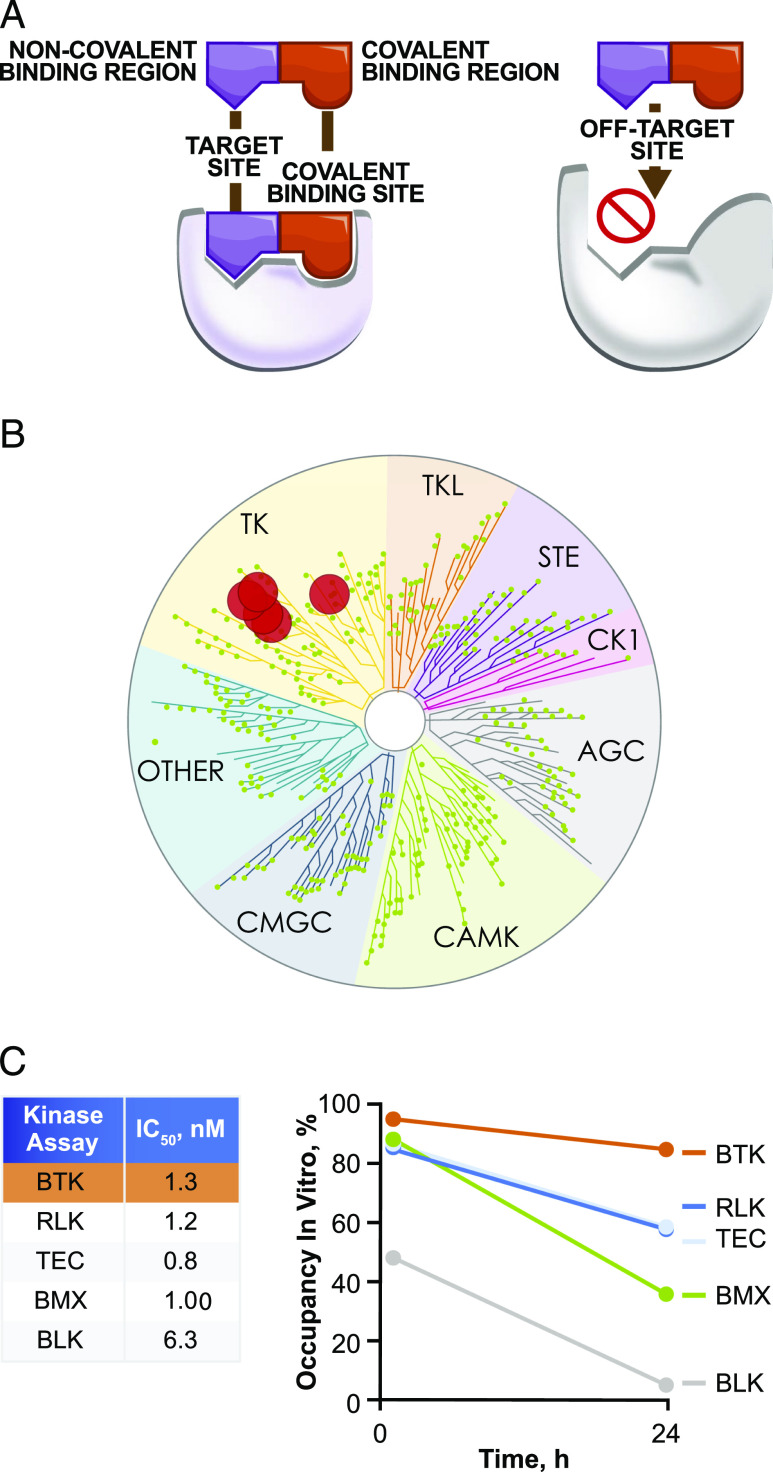
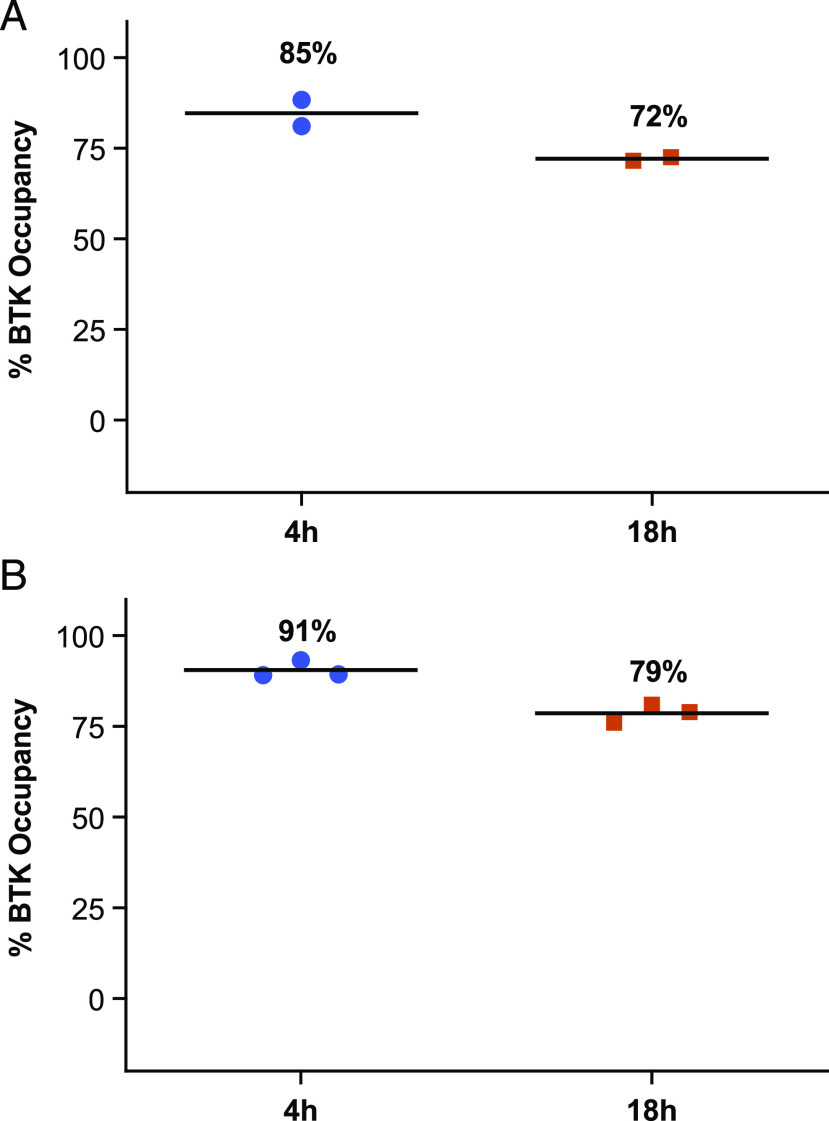
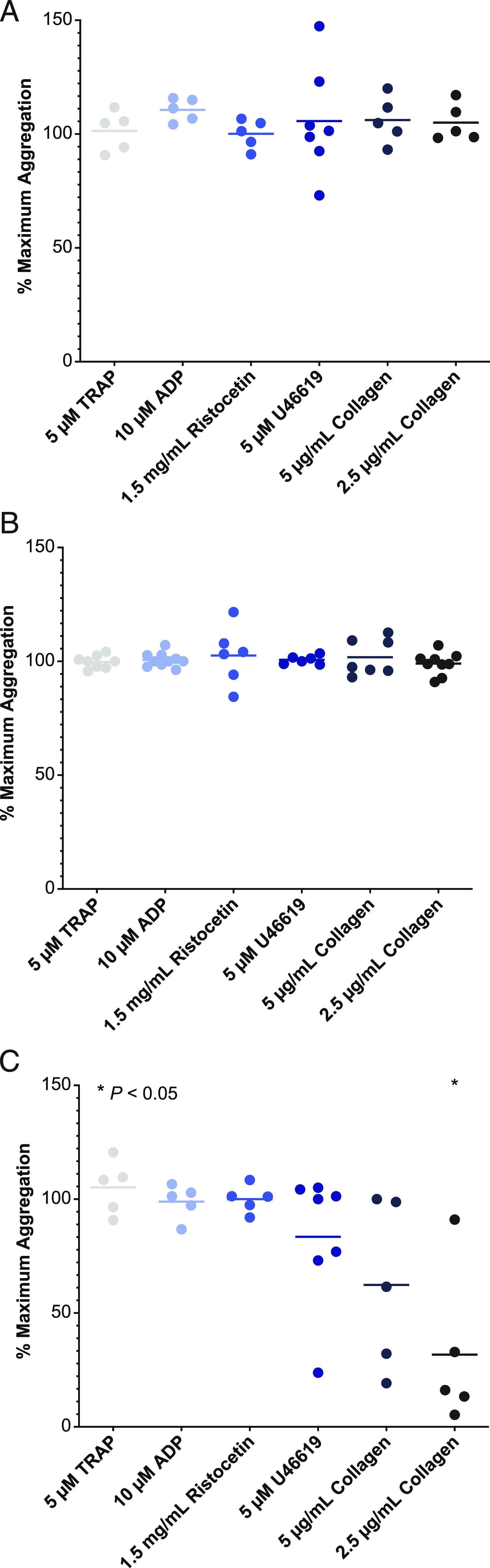
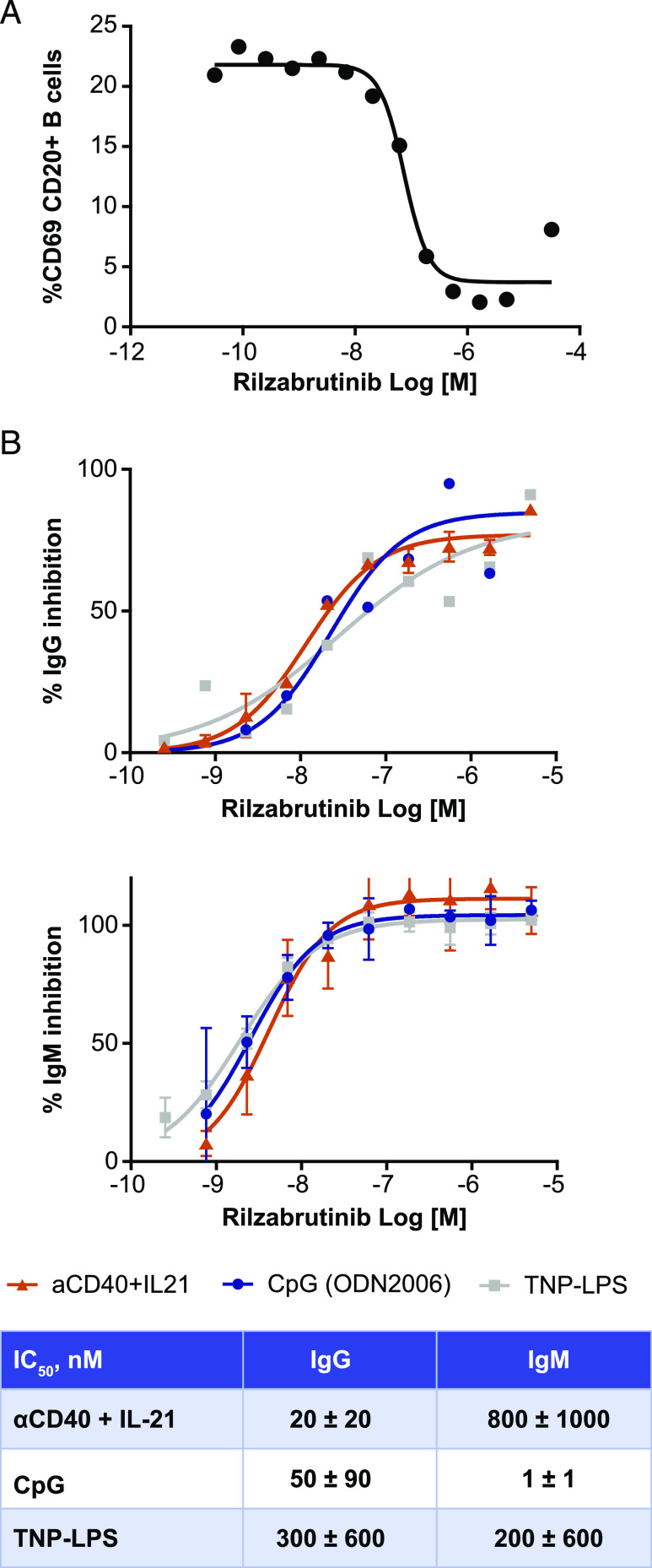
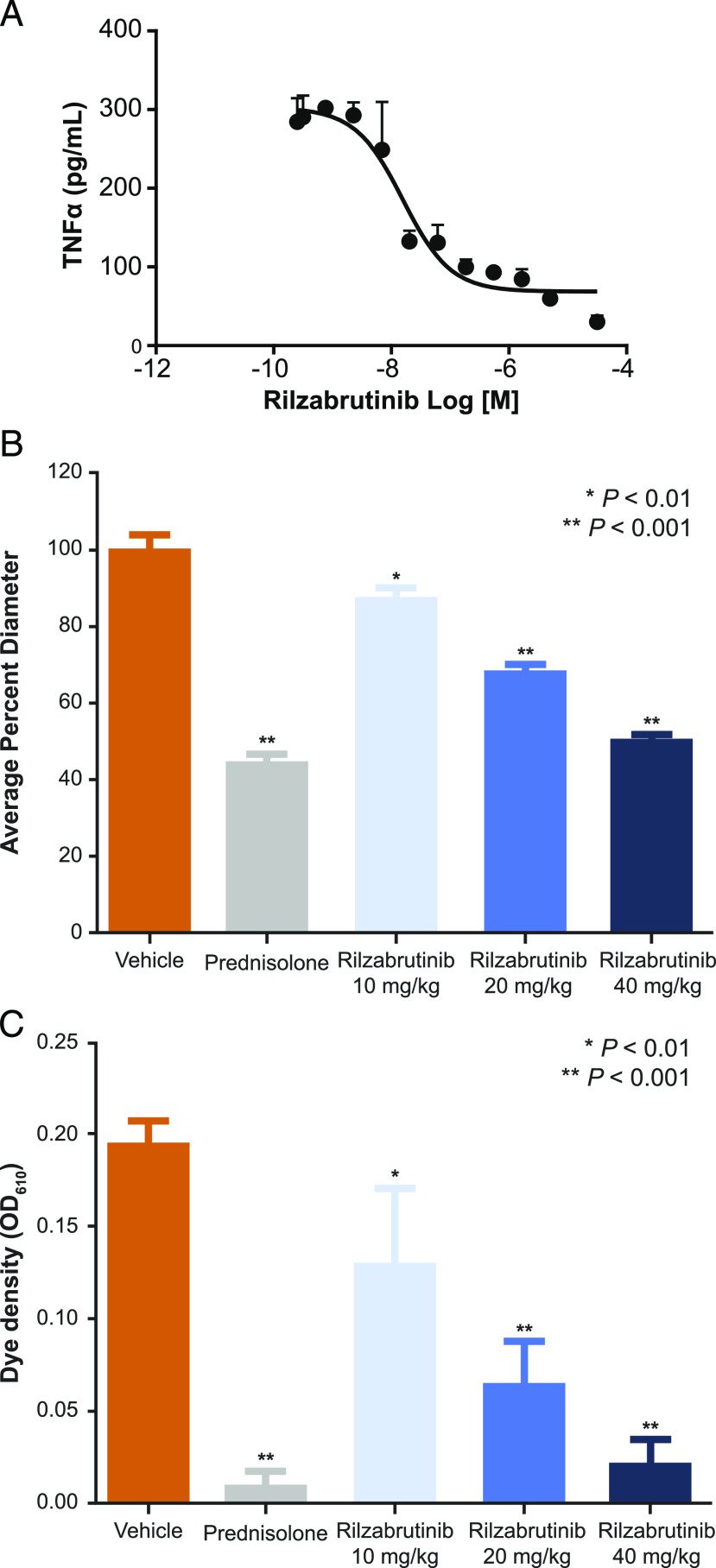
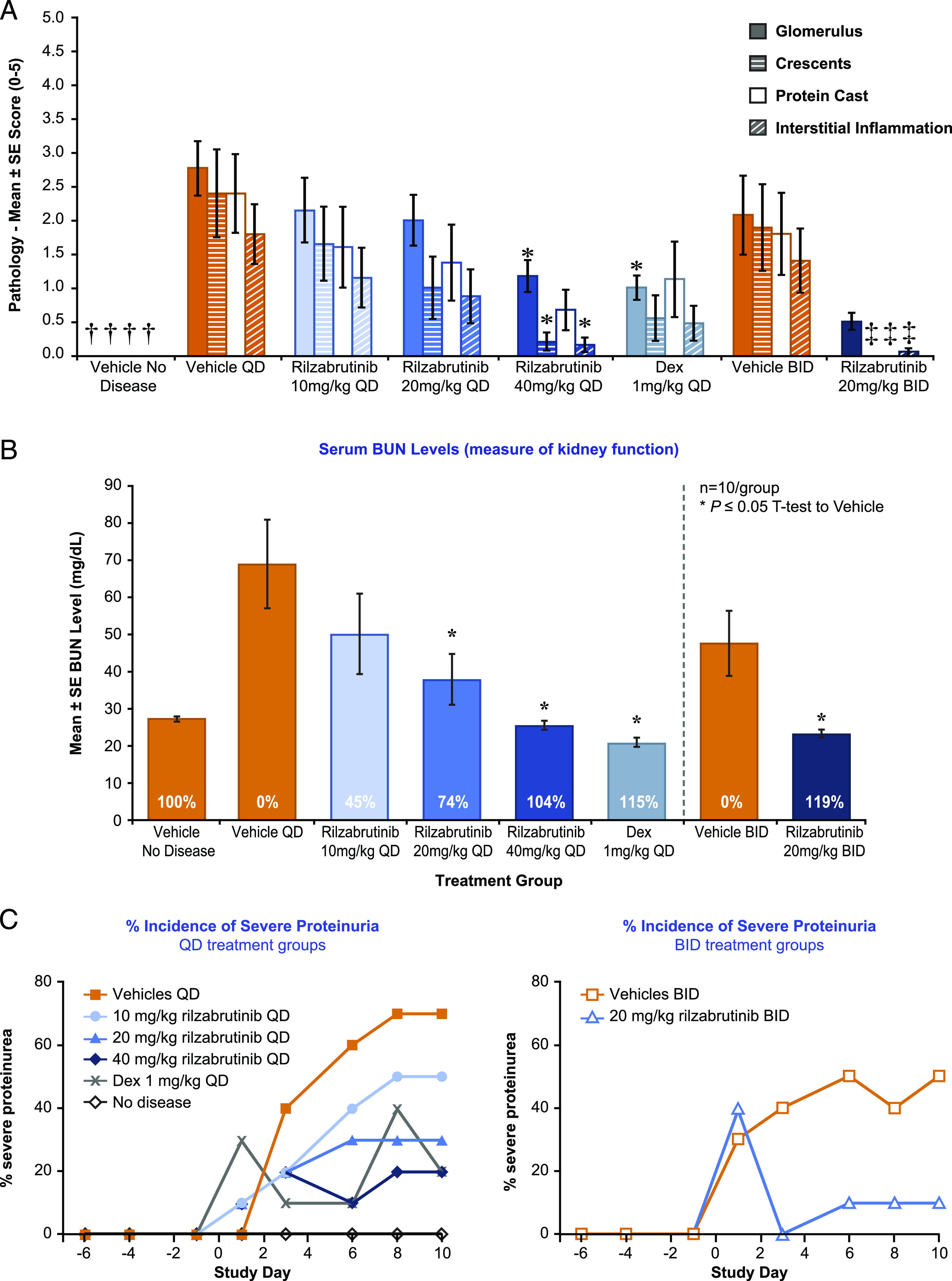
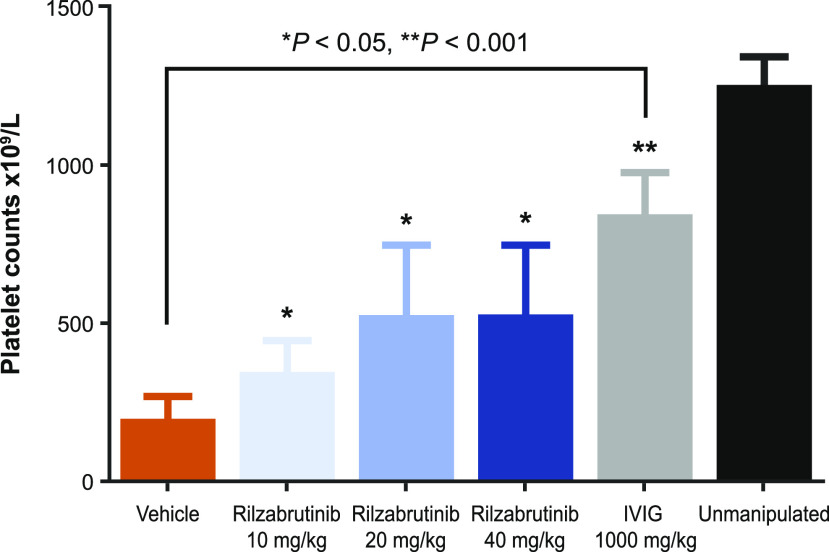
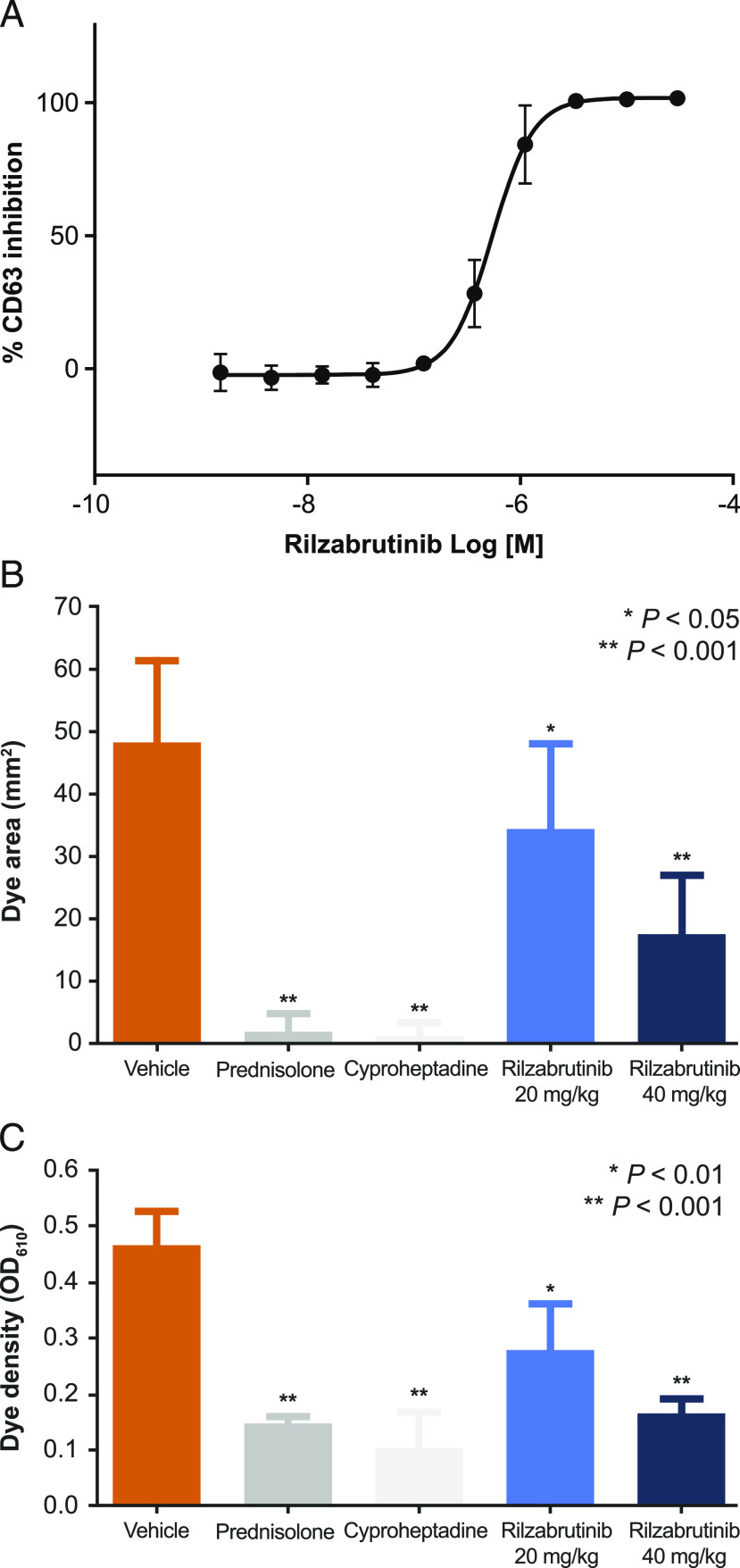
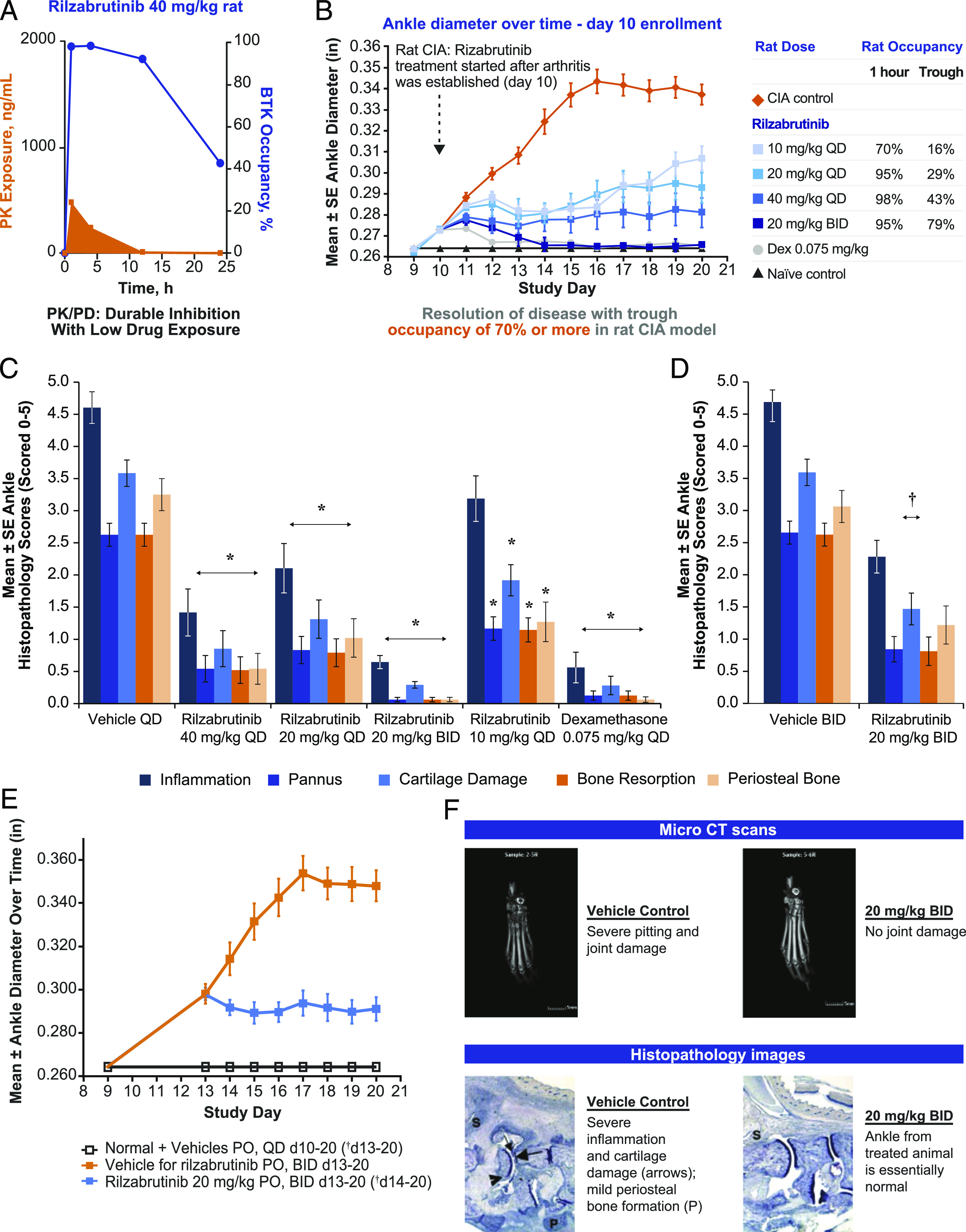
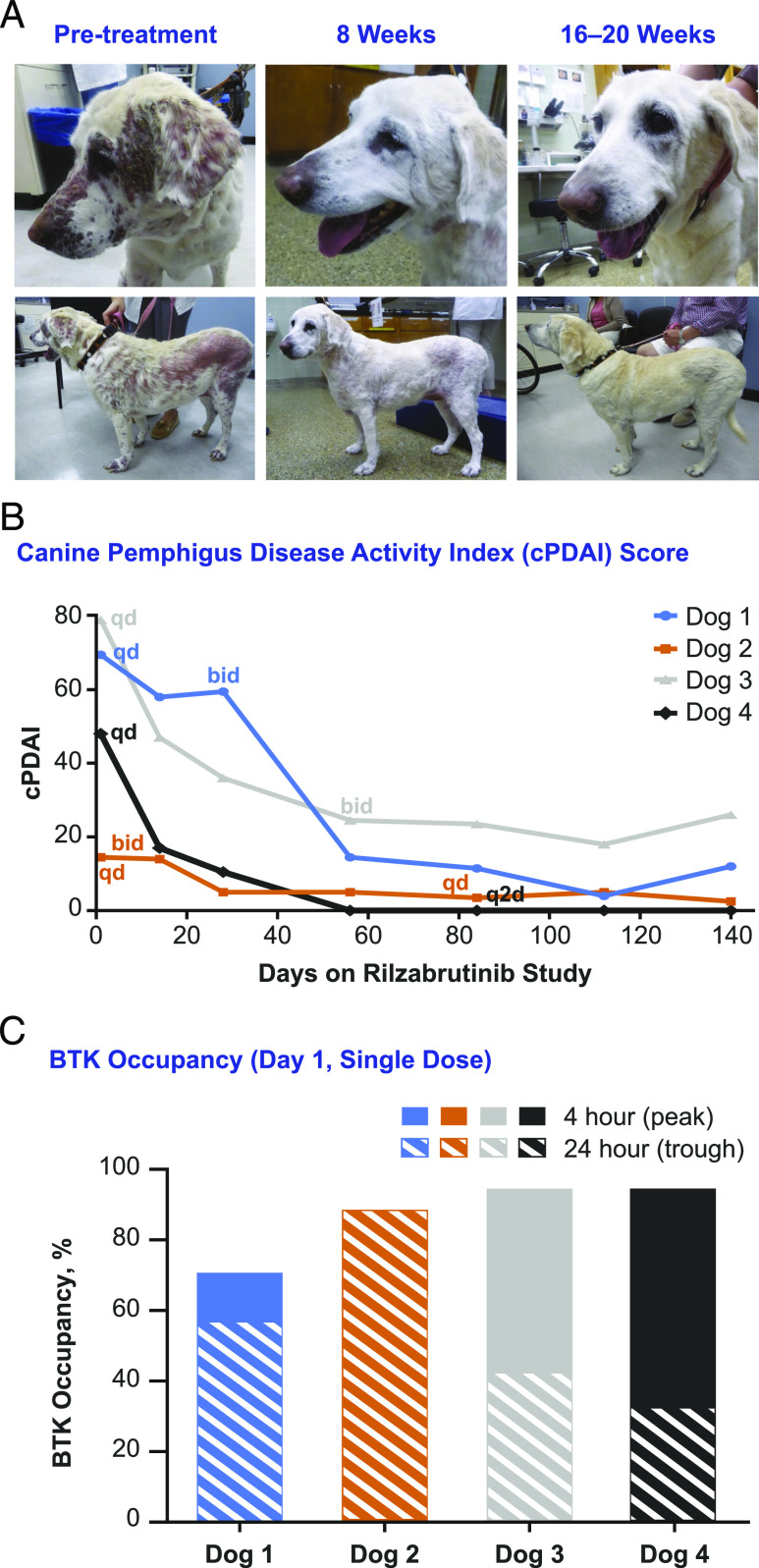
Bruton tyrosine kinase (BTK) is expressed in B cells and innate immune cells, acting as an essential signaling element in multiple immune cell pathways. Selective BTK inhibition has the potential to target multiple immune-mediated disease pathways. Rilzabrutinib is an oral, reversible, covalent BTK inhibitor designed for immune-mediated diseases. We examined the pharmacodynamic profile of rilzabrutinib and its preclinical mechanisms of action. In addition to potent and selective BTK enzyme and cellular activity, rilzabrutinib inhibited activation and inflammatory activities of B cells and innate cells such as macrophages, basophils, mast cells, and neutrophils, without cell death (in human and rodent assay systems). Rilzabrutinib demonstrated dose-dependent improvement of clinical scores and joint pathology in a rat model of collagen-induced arthritis and demonstrated reductions in autoantibody-mediated FcγR signaling in vitro and in vivo, with blockade of rat Arthus reaction, kidney protection in mouse Ab-induced nephritis, and reduction in platelet loss in mouse immune thrombocytopenia. Additionally, rilzabrutinib inhibited IgE-mediated, FcεR-dependent immune mechanisms in human basophils and mast cell-dependent mouse models. In canines with naturally occurring pemphigus, rilzabrutinib treatment resulted in rapid clinical improvement demonstrated by anti-inflammatory effects visible within 2 wk and all animals proceeding to complete or substantial disease control. Rilzabrutinib is characterized by reversible covalent BTK binding, long BTK residence time with low systemic exposure, and multiple mechanistic and biological effects on immune cells. Rilzabrutinib's unique characteristics and promising efficacy and safety profile support clinical development of rilzabrutinib for a broad array of immune-mediated diseases.
Copyright © 2021 by The American Association of Immunologists, Inc.
Conflict of interest statement
C.L.L., J.M.B., T.D.O., M.R.F., Y.X., J.S., J.L., K.A.B., D.M.G., P.A.N. are employees of and receive stock ownership from Principia Biopharma Inc. A.B. is an employee of and receives stock ownership from Precision for Medicine. R.J.H. is an employee of and receives stock ownership from AbbVie. The other authors have no financial conflicts of interest.
Figures
References
-
- Mohamed, A. J., Yu L., Bäckesjö C. M., Vargas L., Faryal R., Aints A., Christensson B., Berglöf A., Vihinen M., Nore B. F., Smith C. I.. 2009. Bruton’s tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol. Rev. 228: 58–73. - PubMed
-
- Rip, J., Van Der Ploeg E. K., Hendriks R. W., Corneth O. B. J.. 2018. The role of Bruton’s tyrosine kinase in immune cell signaling and systemic autoimmunity. Crit. Rev. Immunol. 38: 17–62. - PubMed
-
- Herter, J. M., Margraf A., Volmering S., Correia B. E., Bradshaw J. M., Bisconte A., Hill R. J., Langrish C. L., Lowell C. A., Zarbock A.. 2018. PRN473, an inhibitor of Bruton’s tyrosine kinase, inhibits neutrophil recruitment via inhibition of macrophage antigen-1 signalling. Br. J. Pharmacol. 175: 429–439. - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
