

Tumor methionine metabolism drives T-cell exhaustion in hepatocellular carcinoma
- PMID: 33674593
- PMCID: PMC7935900
- DOI: 10.1038/s41467-021-21804-1
Tumor methionine metabolism drives T-cell exhaustion in hepatocellular carcinoma
Abstract
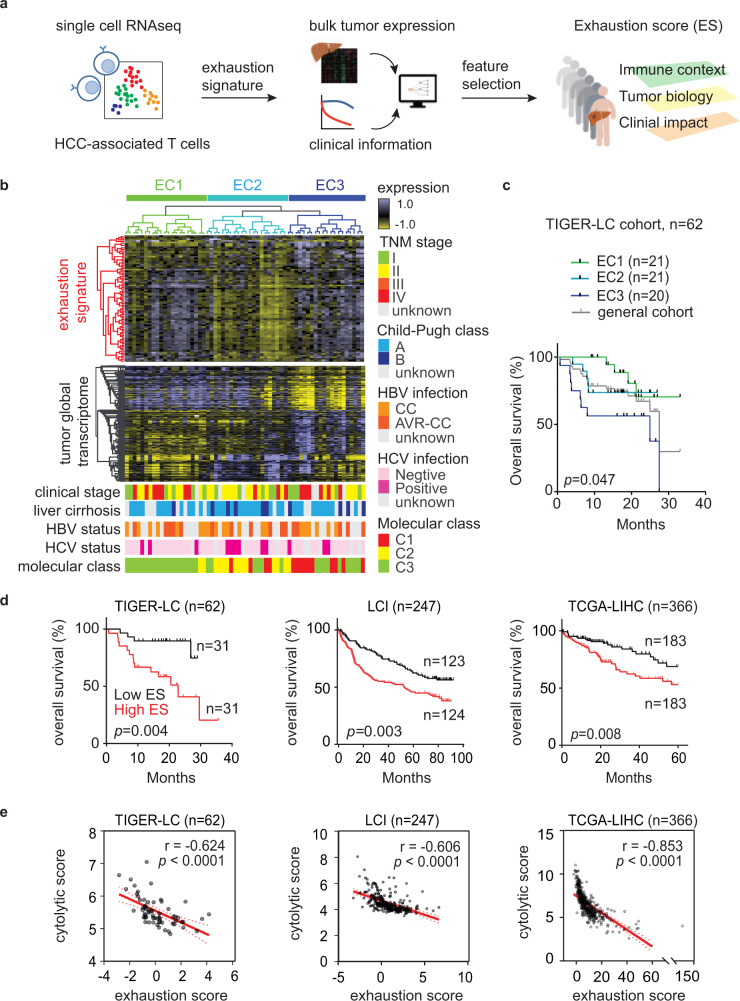
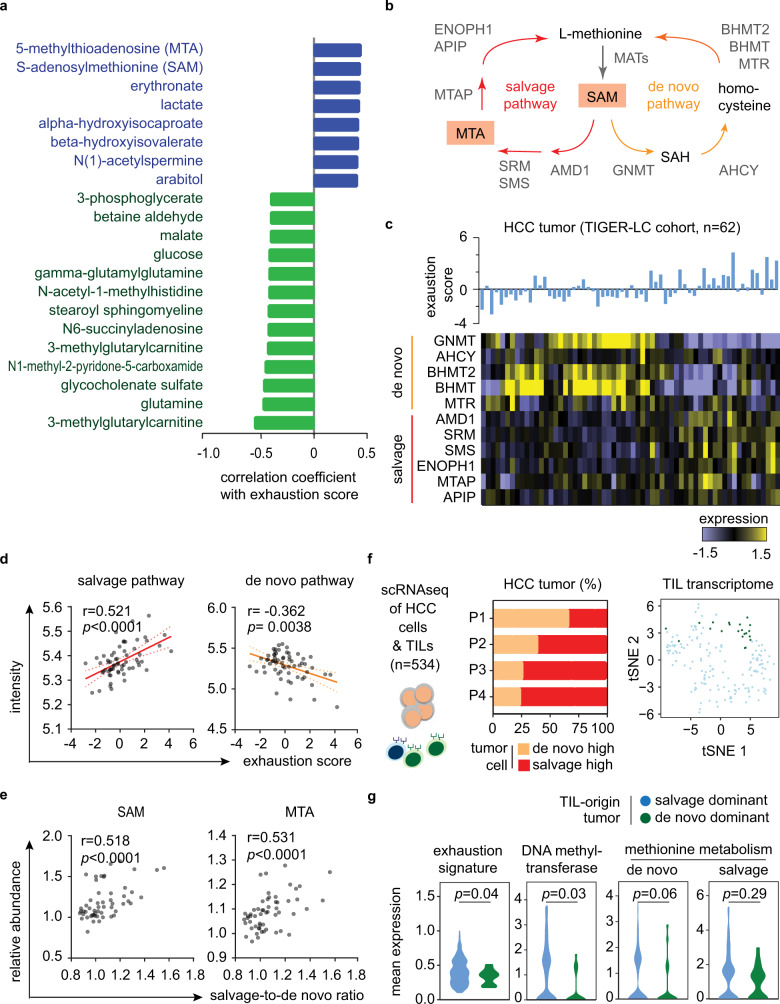
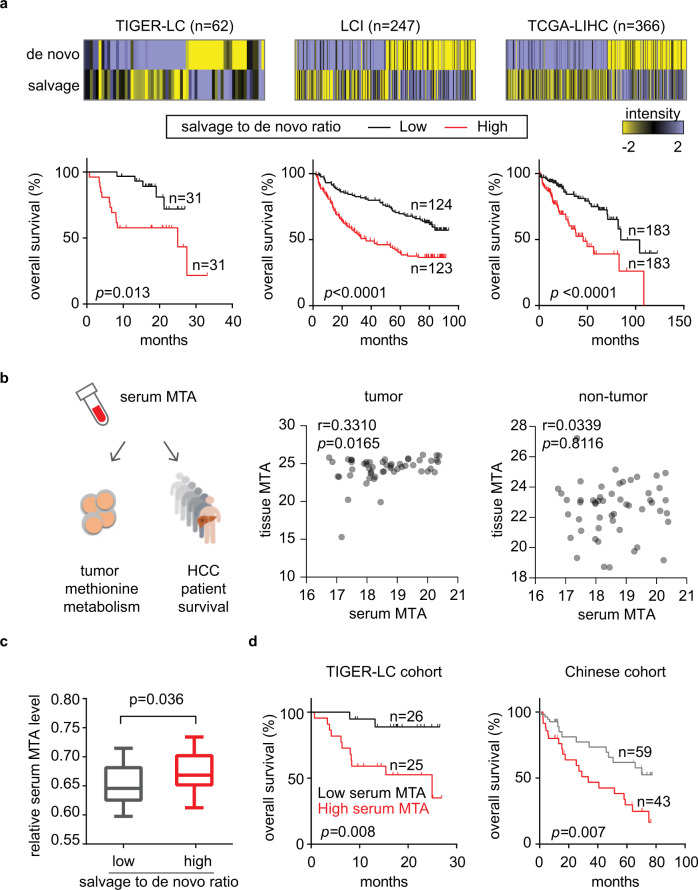
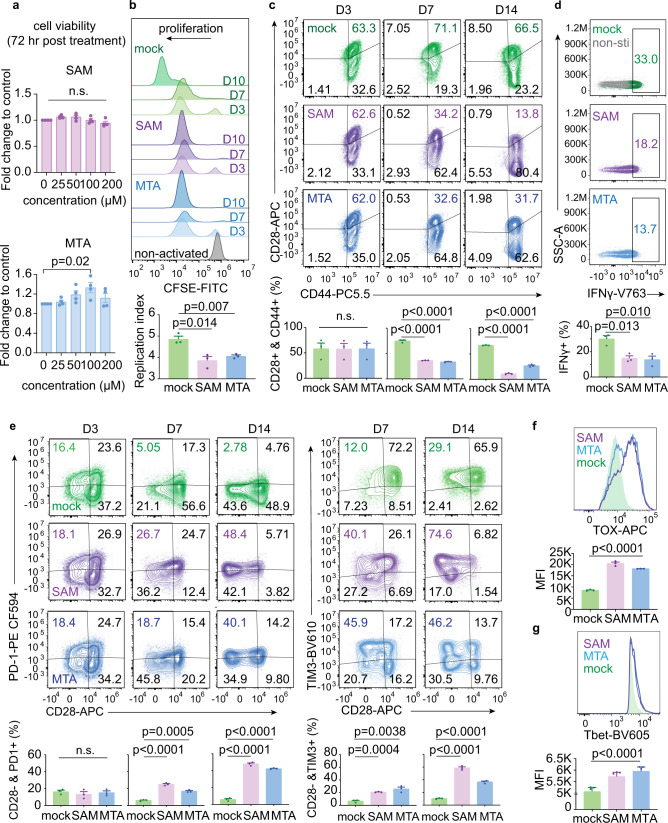
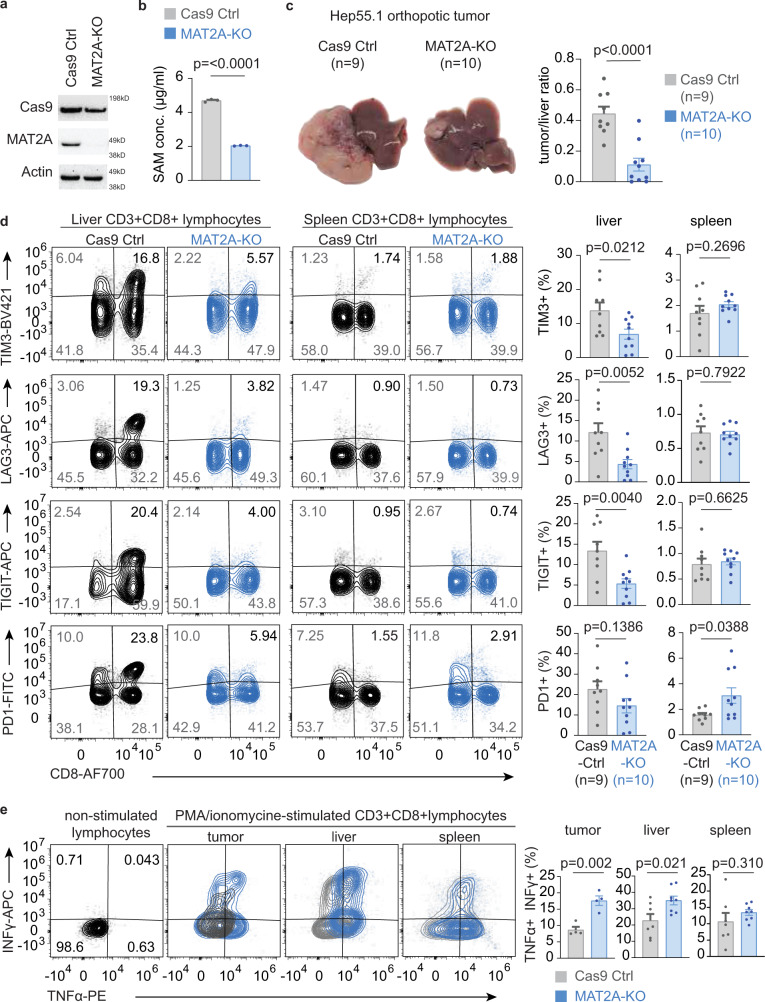
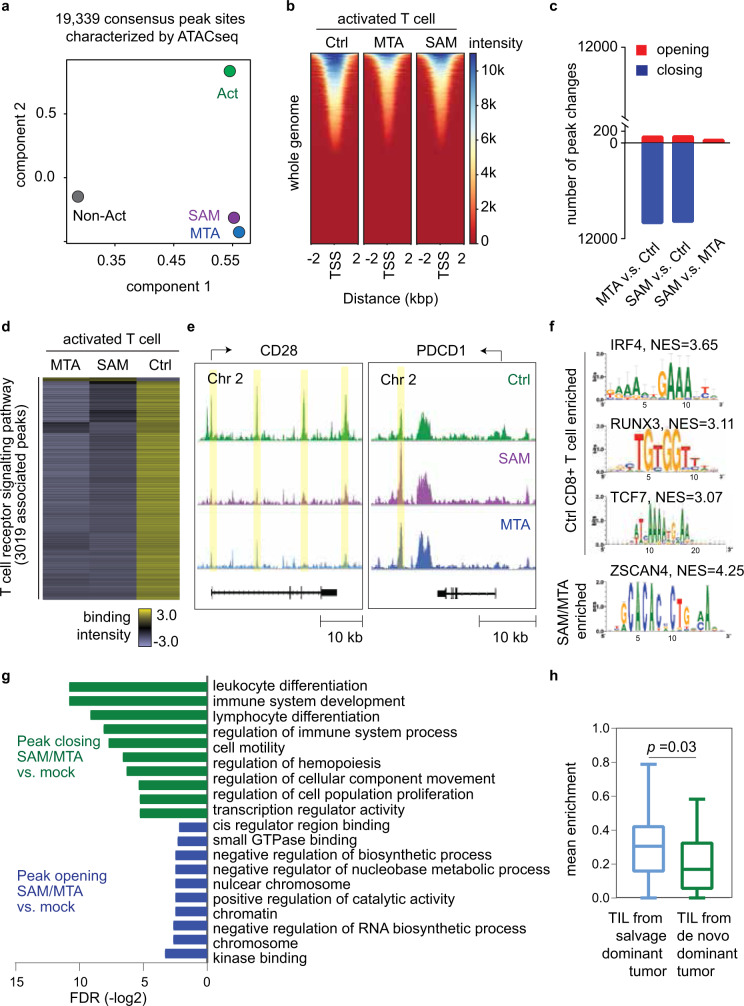
T-cell exhaustion denotes a hypofunctional state of T lymphocytes commonly found in cancer, but how tumor cells drive T-cell exhaustion remains elusive. Here, we find T-cell exhaustion linked to overall survival in 675 hepatocellular carcinoma (HCC) patients with diverse ethnicities and etiologies. Integrative omics analyses uncover oncogenic reprograming of HCC methionine recycling with elevated 5-methylthioadenosine (MTA) and S-adenosylmethionine (SAM) to be tightly linked to T-cell exhaustion. SAM and MTA induce T-cell dysfunction in vitro. Moreover, CRISPR-Cas9-mediated deletion of MAT2A, a key SAM producing enzyme, results in an inhibition of T-cell dysfunction and HCC growth in mice. Thus, reprogramming of tumor methionine metabolism may be a viable therapeutic strategy to improve HCC immunity.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
