

Targeting Bruton tyrosine kinase using non-covalent inhibitors in B cell malignancies
- PMID: 33676527
- PMCID: PMC7937220
- DOI: 10.1186/s13045-021-01049-7
Targeting Bruton tyrosine kinase using non-covalent inhibitors in B cell malignancies
Abstract
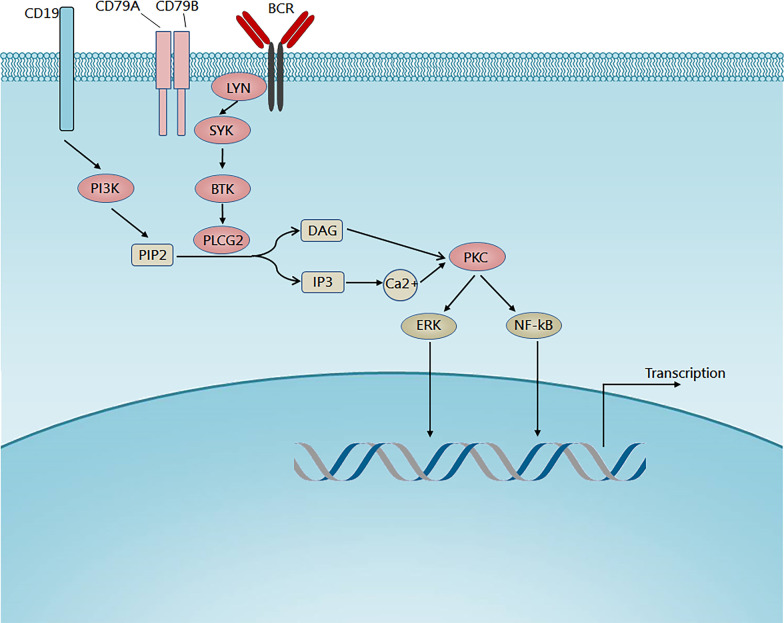
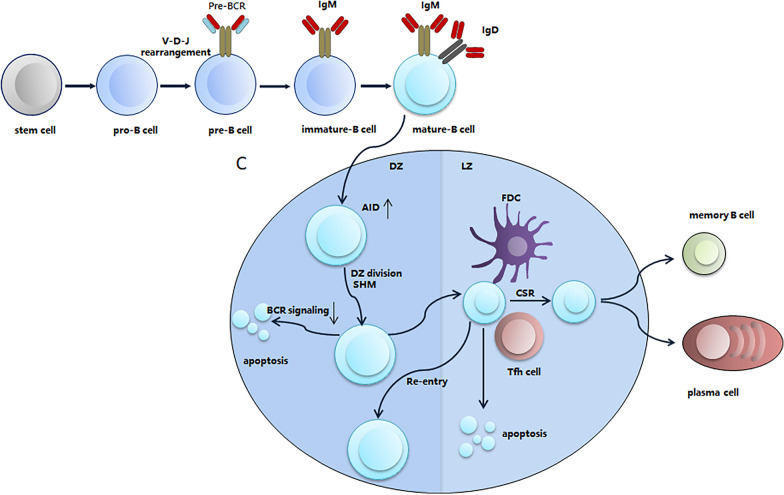
B cell receptor (BCR) signaling is involved in the pathogenesis of B cell malignancies. Activation of BCR signaling promotes the survival and proliferation of malignant B cells. Bruton tyrosine kinase (BTK) is a key component of BCR signaling, establishing BTK as an important therapeutic target. Several covalent BTK inhibitors have shown remarkable efficacy in the treatment of B cell malignancies, especially chronic lymphocytic leukemia. However, acquired resistance to covalent BTK inhibitors is not rare in B cell malignancies. A major mechanism for the acquired resistance is the emergence of BTK cysteine 481 (C481) mutations, which disrupt the binding of covalent BTK inhibitors. Additionally, adverse events due to the off-target inhibition of kinases other than BTK by covalent inhibitors are common. Alternative therapeutic options are needed if acquired resistance or intolerable adverse events occur. Non-covalent BTK inhibitors do not bind to C481, therefore providing a potentially effective option to patients with B cell malignancies, including those who have developed resistance to covalent BTK inhibitors. Preliminary clinical studies have suggested that non-covalent BTK inhibitors are effective and well-tolerated. In this review, we discussed the rationale for the use of non-covalent BTK inhibitors and the preclinical and clinical studies of non-covalent BTK inhibitors in B cell malignancies.
Keywords: B cell malignancies; BTK; C481 mutations; Ibrutinib; Non-covalent inhibitors.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
