

Application of single-cell transcriptomics to kinetoplastid research
- PMID: 33678213
- PMCID: PMC8311972
- DOI: 10.1017/S003118202100041X
Application of single-cell transcriptomics to kinetoplastid research
Abstract
Kinetoplastid parasites are responsible for both human and animal diseases across the globe where they have a great impact on health and economic well-being. Many species and life cycle stages are difficult to study due to limitations in isolation and culture, as well as to their existence as heterogeneous populations in hosts and vectors. Single-cell transcriptomics (scRNA-seq) has the capacity to overcome many of these difficulties, and can be leveraged to disentangle heterogeneous populations, highlight genes crucial for propagation through the life cycle, and enable detailed analysis of host–parasite interactions. Here, we provide a review of studies that have applied scRNA-seq to protozoan parasites so far. In addition, we provide an overview of sample preparation and technology choice considerations when planning scRNA-seq experiments, as well as challenges faced when analysing the large amounts of data generated. Finally, we highlight areas of kinetoplastid research that could benefit from scRNA-seq technologies.
Keywords: bioinformatics; gene expression; kinetoplastid; parasitology; single-cell transcriptomics.
Conflict of interest statement
The authors declare there are no conflicts of interest.
Figures
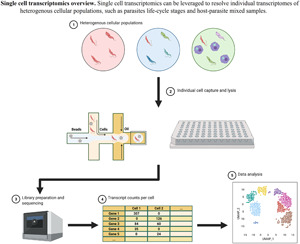
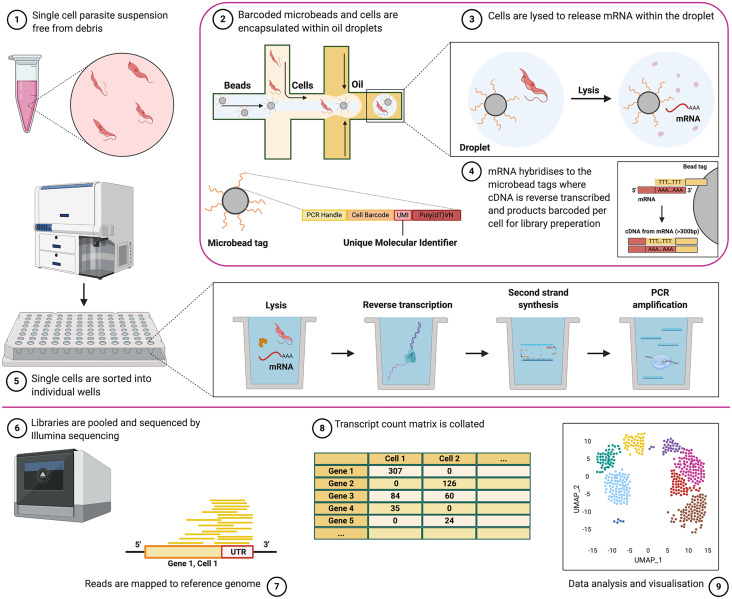


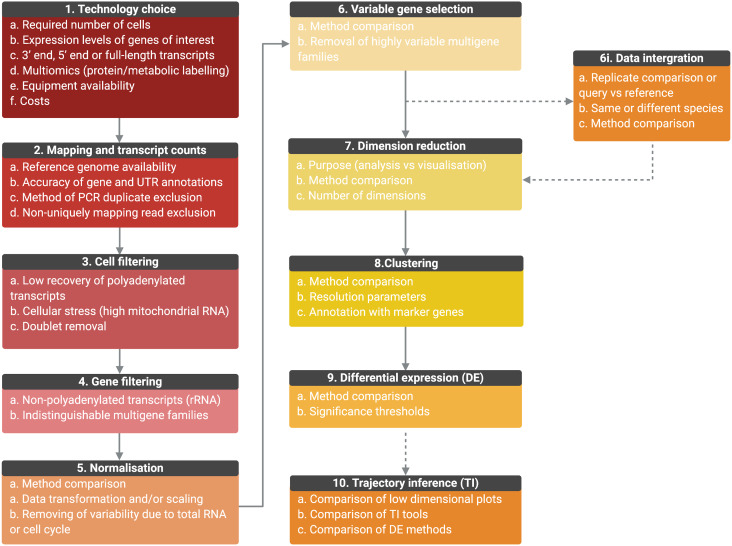
References
-
- 10x Genomics (2020a) Cell ranger – Software overview. Available at: https://support.10xgenomics.com/single-cell-gene-expression/software/ove... (Accessed: 15 December 2020).
-
- 10x Genomics (2020b) Chromium Next GEM Single Cell 5′ v2. Available at: https://support.10xgenomics.com/single-cell-vdj/library-prep/doc/technic... (Accessed: 15 December 2020).
-
- Alfituri OA, Quintana JF, MacLeod A, Garside P, Benson RA, Brewer JM, Mabbott NA, Morrison LJ and Capewell P (2020) To the skin and beyond: the immune response to African trypanosomes as they enter and exit the vertebrate host. Frontiers in Immunology 11, e1250. doi: 10.3389/fimmu.2020.01250 - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
