

Case Report: A Novel Synonymous ARPC1B Gene Mutation Causes a Syndrome of Combined Immunodeficiency, Asthma, and Allergy With Significant Intrafamilial Clinical Heterogeneity
- PMID: 33679784
- PMCID: PMC7933039
- DOI: 10.3389/fimmu.2021.634313
Case Report: A Novel Synonymous ARPC1B Gene Mutation Causes a Syndrome of Combined Immunodeficiency, Asthma, and Allergy With Significant Intrafamilial Clinical Heterogeneity
Abstract
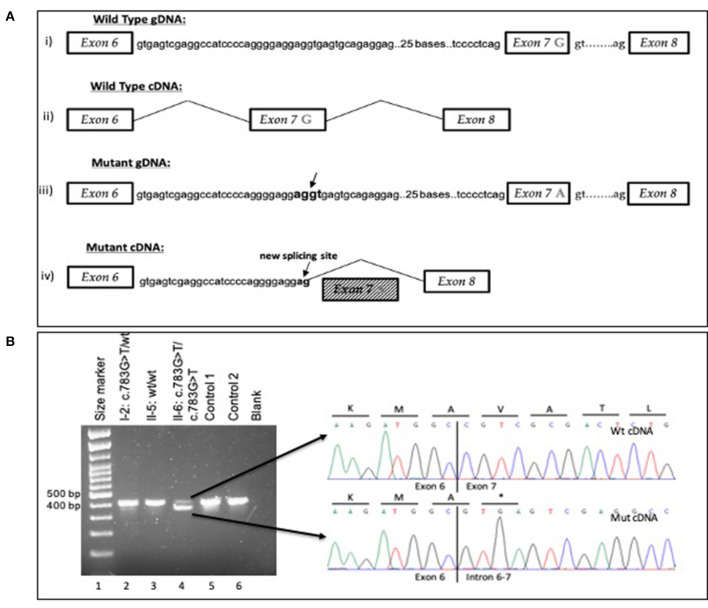
Recently, a novel syndrome of combined immune deficiency, infections, allergy, and inflammation has been attributed to mutations in the gene encoding actin-related protein 2/3 complex subunit 1B (ARPC1B), which is a key molecule driving the dynamics of the cytoskeleton. Homozygous mutations in the ARPC1B gene have been found to result in the disruption of the protein structure and cause an autosomal recessive syndrome of combined immune deficiency, impaired T-cell migration and proliferation, increased levels of immunoglobulin E (IgE) and immunoglobulin A (IgA), and thrombocytopenia. To date, only a few individuals have been diagnosed with the ARPC1B deficiency syndrome worldwide. In this case series, we report the wide spectrum of phenotype in 3 siblings of a consanguineous family from Afghanistan with a novel homozygous synonymous pathogenic variant c.783G>A, p. (Ala261Ala) of the ARPC1B gene that causes a similar syndrome but no thrombocytopenia. Targeted RNA studies demonstrated that the variant affects the splicing process of mRNA, resulting in a marked reduction of the levels of primary (normal) RNA transcript of the ARPC1B gene in the affected patients and likely premature termination from the abnormally spliced mRNA. The next generation sequencing (NGS) studies facilitated the diagnosis of this rare combined immunodeficiency and led to the decision to treat the affected patients with hematopoietic cell transplant (HCT) from an human leukocyte antigen (HLA)-matched healthy sibling.
Keywords: Arpc1b; allergy; case report; combined immune deficiency; immunodeficiency; inborn errors of immunity.
Copyright © 2021 Papadatou, Marinakis, Botsa, Tzanoudaki, Kanariou, Orfanou, Kanaka-Gantenbein, Traeger-Synodinos and Spoulou.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous