

Metabolic Alteration in Hepatocellular Carcinoma: Mechanism of Lipid Accumulation in Well-Differentiated Hepatocellular Carcinoma
- PMID: 33681091
- PMCID: PMC7910064
- DOI: 10.1155/2021/8813410
Metabolic Alteration in Hepatocellular Carcinoma: Mechanism of Lipid Accumulation in Well-Differentiated Hepatocellular Carcinoma
Abstract
Objective: Metabolic alteration is widely considered as one of the hallmarks of cancer. Hepatocellular carcinoma (HCC) presents a unique pathological feature in which lipid accumulation is common in well-differentiated HCC and rare in poorly differentiated HCC; however, the underlying mechanism remains unclear.
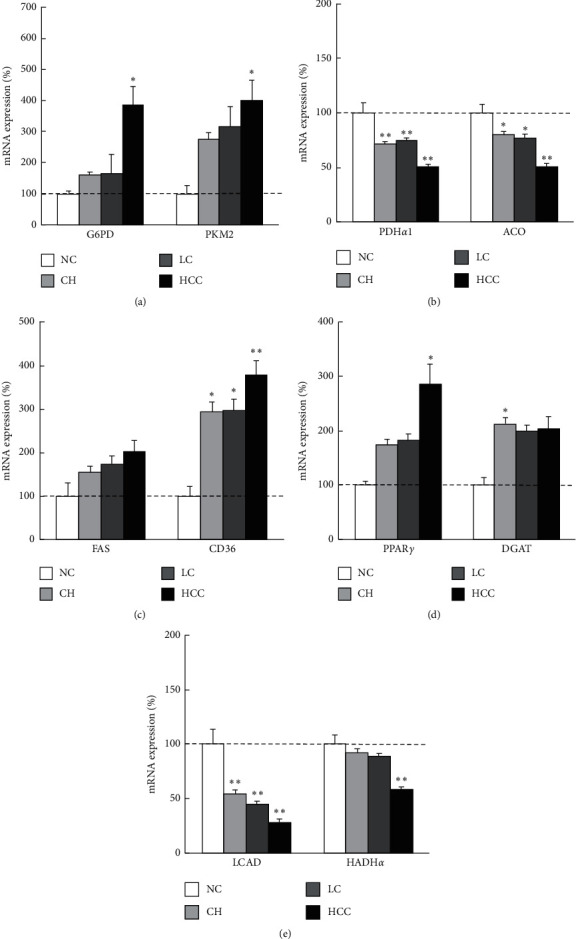
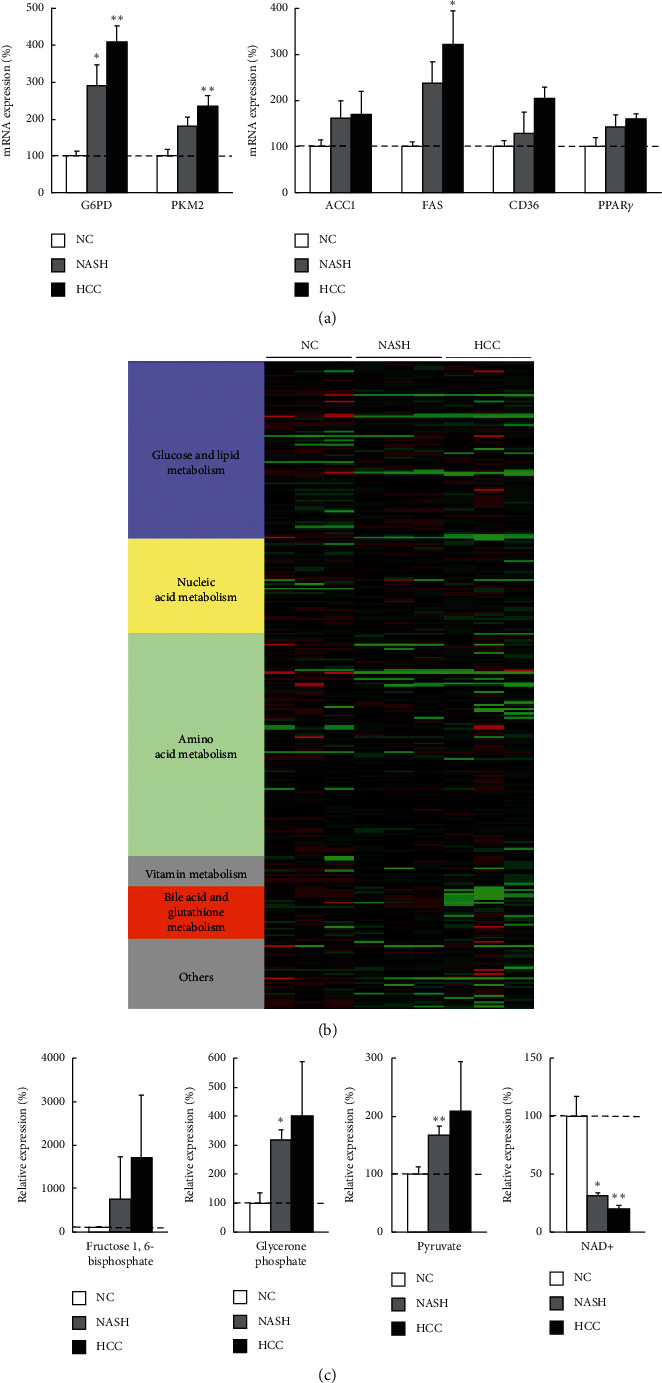
Methods: Tissue samples were obtained from 103 HCC patients who had undergone hepatic resection and 12 living donors of liver transplantation. We evaluated metabolic gene expressions in cancer tissues as well as background noncancer tissues and compared the expressions by the degree of cancer differentiation and by liver disease states. Besides, the metabolomics was evaluated and integrated to gene expressions in nonalcoholic steatohepatitis (NASH)-HCC model mice.
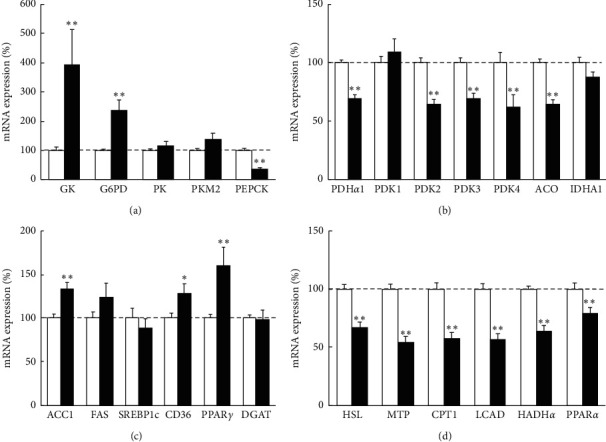
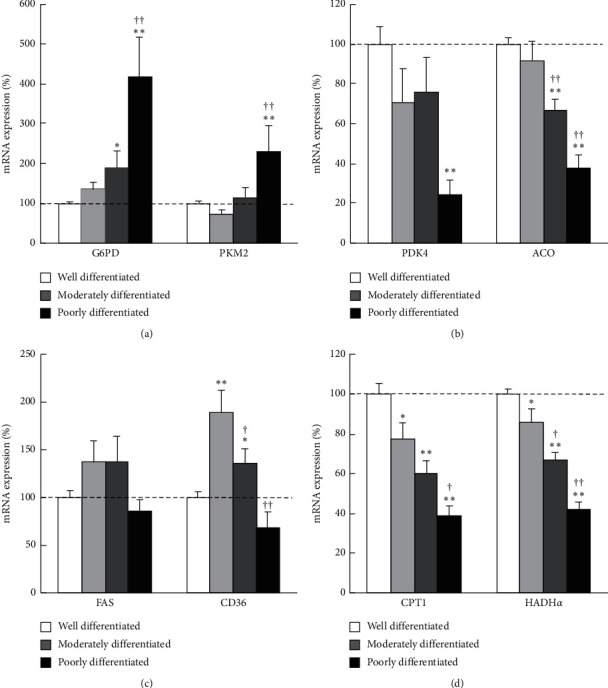
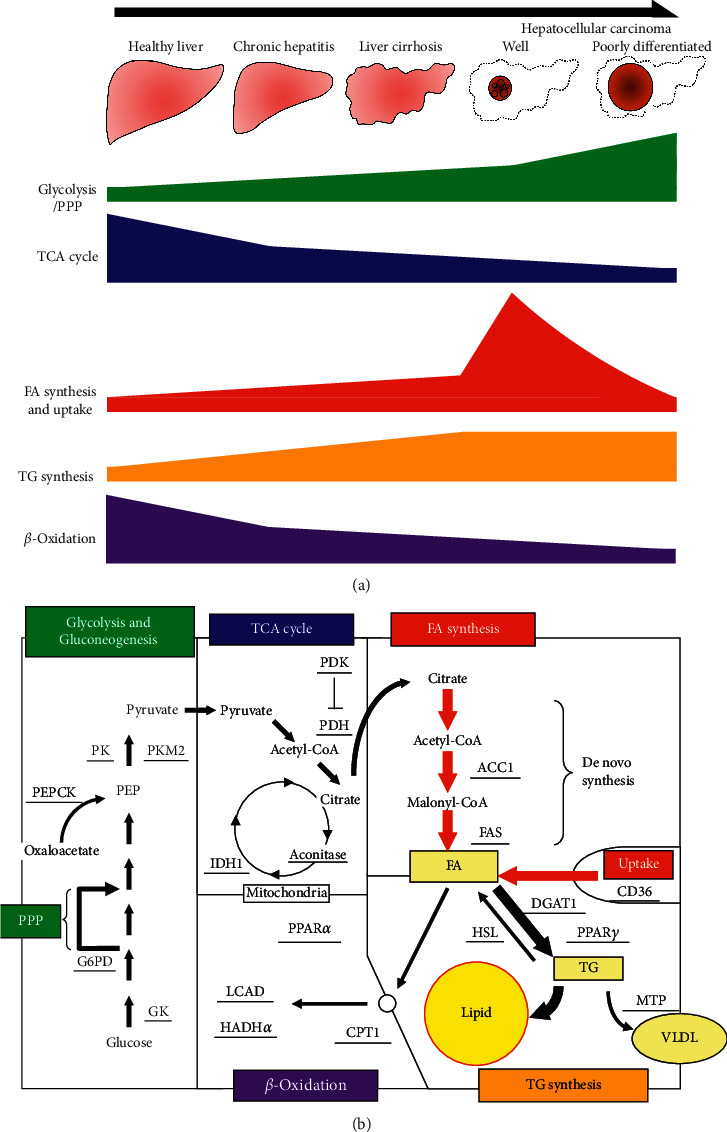
Results: In cancer tissues, the expression levels of enzymes related to glycolysis, pentose phosphate pathway (PPP), and fatty acid (FA) synthesis were increased and that of tricarboxylic acid (TCA) cycle and β-oxidation were suppressed. Same metabolic alterations were observed in noncancer tissue as the liver disease progresses from healthy liver to chronic hepatitis, cirrhosis, and HCC. Similar alterations of metabolic genes were detected in NASH-HCC mice, which were consistent with the results of metabolomics. As the degree of cancer differentiation decreased, glycolysis and PPP were accelerated; however, FA synthesis and uptake were diminished.
Conclusions: The metabolic alterations including glycolysis, PPP, TCA cycle, and β-oxidation became more prominent as liver disease progresses from normal, chronic hepatitis, cirrhosis, well-, moderately, and poorly differentiated HCC. FA synthesis and uptake were highest in well-differentiated HCC, which could explain the lipid accumulation.
Copyright © 2021 Hideo Suzuki et al.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
