

Clinical profile and treatment outcome of epilepsy syndromes in children: A hospital-based study in Eastern Nepal
- PMID: 33681663
- PMCID: PMC7918298
- DOI: 10.1002/epi4.12470
Clinical profile and treatment outcome of epilepsy syndromes in children: A hospital-based study in Eastern Nepal
Abstract
Objective: It is often difficult to diagnose epilepsy syndromes in resource-limited settings. This study was aimed to investigate the prospect of ascertaining the diagnosis, clinical profile, and treatment outcomes of epilepsy syndromes (ESs) among children in a resource-limited setting.
Methods: This was a descriptive study done from 01/07/2009 to 15/06/2017 among children (1-17 years of age) with unprovoked seizures presenting to the pediatric neurology clinic of a university hospital in eastern Nepal. Diagnosis, classification, and treatment of seizures were based upon International League Against Epilepsy guidelines.
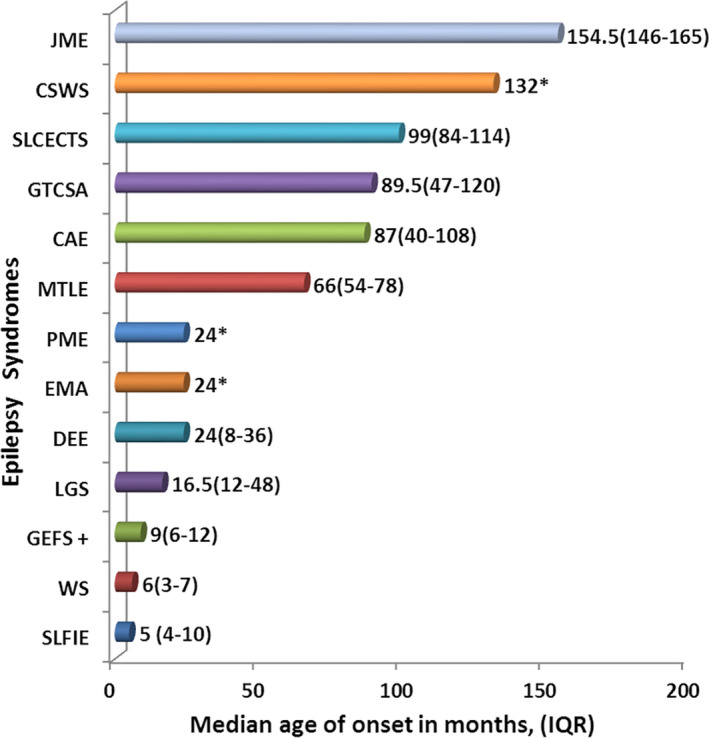
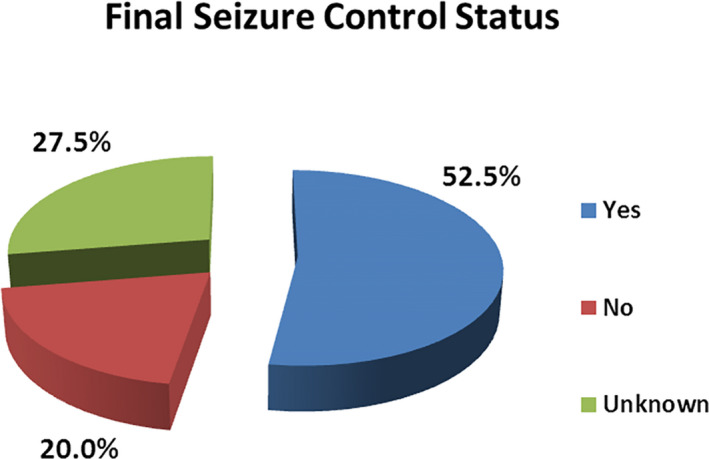
Results: Of 768 children with unprovoked seizures, 120 (15.6%) were diagnosed as ES. The age of onset of seizure was unique for each ES. Developmental delay and cerebral palsy were present in 47.5% and 28.3% children, respectively. Common ESs were West syndrome (WS)-26.7%, generalized tonic-clonic seizures alone (GTCSA)-21.7%, self-limited childhood epilepsy with centrotemporal spikes (SLCECTS)-12.5%, childhood absence epilepsy (CAE)-10.0%, Lennox-Gastaut syndrome (LGS)-10.0%, other developmental and epileptic encephalopathies (DEE)-5.8%, self-limited familial infantile epilepsy (SLFIE)-4.2%, and juvenile myoclonic epilepsy (JME)-3.3%. Among children with known outcomes (87/120), overall response to pharmacotherapy and to monotherapy was observed in 72.4% (63/87) and 57.5% (50/87) children, respectively. All children with GTCSA, SLFIE, genetic epilepsy with febrile seizure plus (GEFS+), CAE, SLCECTS, and JME responded to pharmacotherapy and they had normal computerized tomography scans of the brain. Seizures were largely pharmaco-resistant in progressive myoclonus epilepsy (PME)-100.0%, LGS-73.0%, WS-52.0%, and other DEEs-40%.
Significance: A reasonable proportion (15.6%) of unprovoked seizures could be classified into specific ES despite limited diagnostic resources. WS was the most common ES. GTCSA, SLCECTS, CAE, and LGS were other common ESs. GTCSA, SLFIE, CAE, SLCECTS, GEFS+, and JME were largely pharmaco-responsive. PME, WS, and LGS were relatively pharmaco-resistant. Electro-clinical diagnosis of certain ES avoids the necessity of neuroimaging.
Keywords: child; epilepsy; epileptic syndromes; seizure; treatment outcome.
© 2021 The Authors. Epilepsia Open published by Wiley Periodicals LLC on behalf of International League Against Epilepsy.
Conflict of interest statement
The authors declare that they have no competing interests. This research received no specific grant from any funding agency in the public, commercial, or not‐for‐profit sectors.
Figures
Similar articles
-
Evolution and course of early life developmental encephalopathic epilepsies: Focus on Lennox-Gastaut syndrome.Epilepsia. 2018 Nov;59(11):2096-2105. doi: 10.1111/epi.14569. Epub 2018 Sep 26. Epilepsia. 2018. PMID: 30255934 Free PMC article.
-
Late infantile epileptic encephalopathy: A distinct developmental and epileptic encephalopathy syndrome.Epileptic Disord. 2024 Feb;26(1):98-108. doi: 10.1002/epd2.20185. Epub 2024 Jan 3. Epileptic Disord. 2024. PMID: 38100275
-
Treatment of pediatric epilepsy: European expert opinion, 2007.Epileptic Disord. 2007 Dec;9(4):353-412. doi: 10.1684/epd.2007.0144. Epileptic Disord. 2007. PMID: 18077226 Review.
-
Diagnosis switching and outcomes in a cohort of patients with potential epilepsy with myoclonic-atonic seizures.Epilepsy Res. 2018 Nov;147:95-101. doi: 10.1016/j.eplepsyres.2018.09.011. Epub 2018 Sep 24. Epilepsy Res. 2018. PMID: 30286391 Free PMC article.
-
Epilepsy: Epileptic Syndromes and Treatment.Neurol Clin. 2021 Aug;39(3):779-795. doi: 10.1016/j.ncl.2021.04.002. Neurol Clin. 2021. PMID: 34215386 Review.
Cited by
-
Pattern and etiology of early childhood epilepsy: An Experience at a tertiary care University Center.Neurosciences (Riyadh). 2022 Oct;27(4):244-250. doi: 10.17712/nsj.2022.4.20220001. Neurosciences (Riyadh). 2022. PMID: 36252977 Free PMC article.
-
Classifying etiology of infantile spasms syndrome in resource-limited settings: A study from the South Asian region.Epilepsia Open. 2021 Dec;6(4):736-747. doi: 10.1002/epi4.12548. Epub 2021 Oct 25. Epilepsia Open. 2021. PMID: 34653320 Free PMC article.
-
Clinical Characteristics, Treatment Outcome and Associated Factors of Epilepsy Among Children at Hospitals of North-West Ethiopia.Pediatric Health Med Ther. 2023 Oct 31;14:385-404. doi: 10.2147/PHMT.S436022. eCollection 2023. Pediatric Health Med Ther. 2023. PMID: 37927397 Free PMC article.
References
-
- Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde BW, et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51(4):676–85. - PubMed
-
- Proposal for revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia 1989;30(4):389–99. - PubMed
-
- Fisher RS, Cross JH, D’Souza C, French JA, Haut SR, Higurashi N, et al. Instruction manual for the ILAE 2017 operational classification of seizure types. Epilepsia. 2017;58(4):531–42. - PubMed
-
- Duchowny M, Harvey AS. Pediatric epilepsy syndromes: an update and critical review. Epilepsia. 1996;37(Suppl 1):S26–40. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
