

Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities
- PMID: 33681822
- PMCID: PMC7935068
- DOI: 10.1038/s43018-020-00159-4
Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities
Abstract
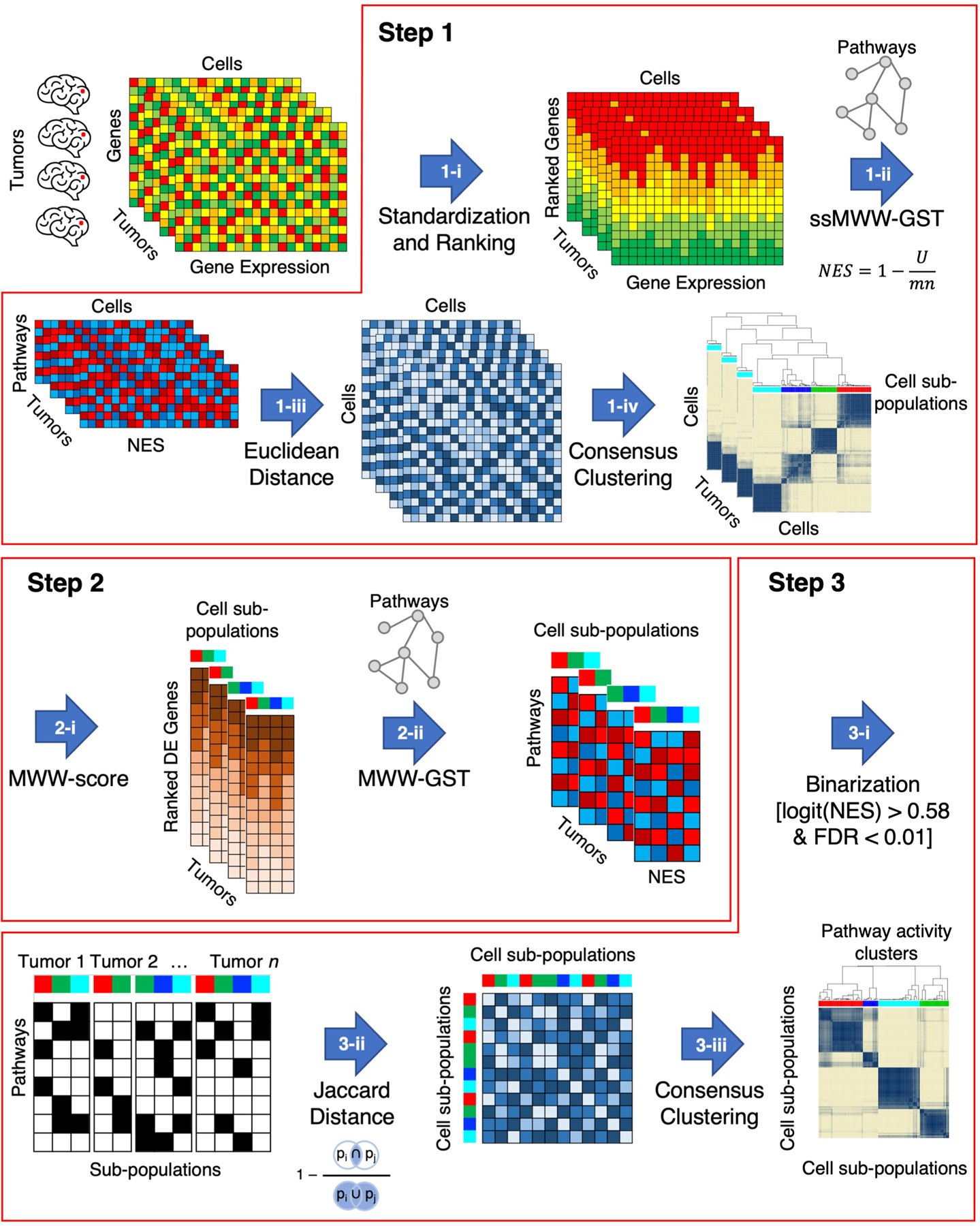
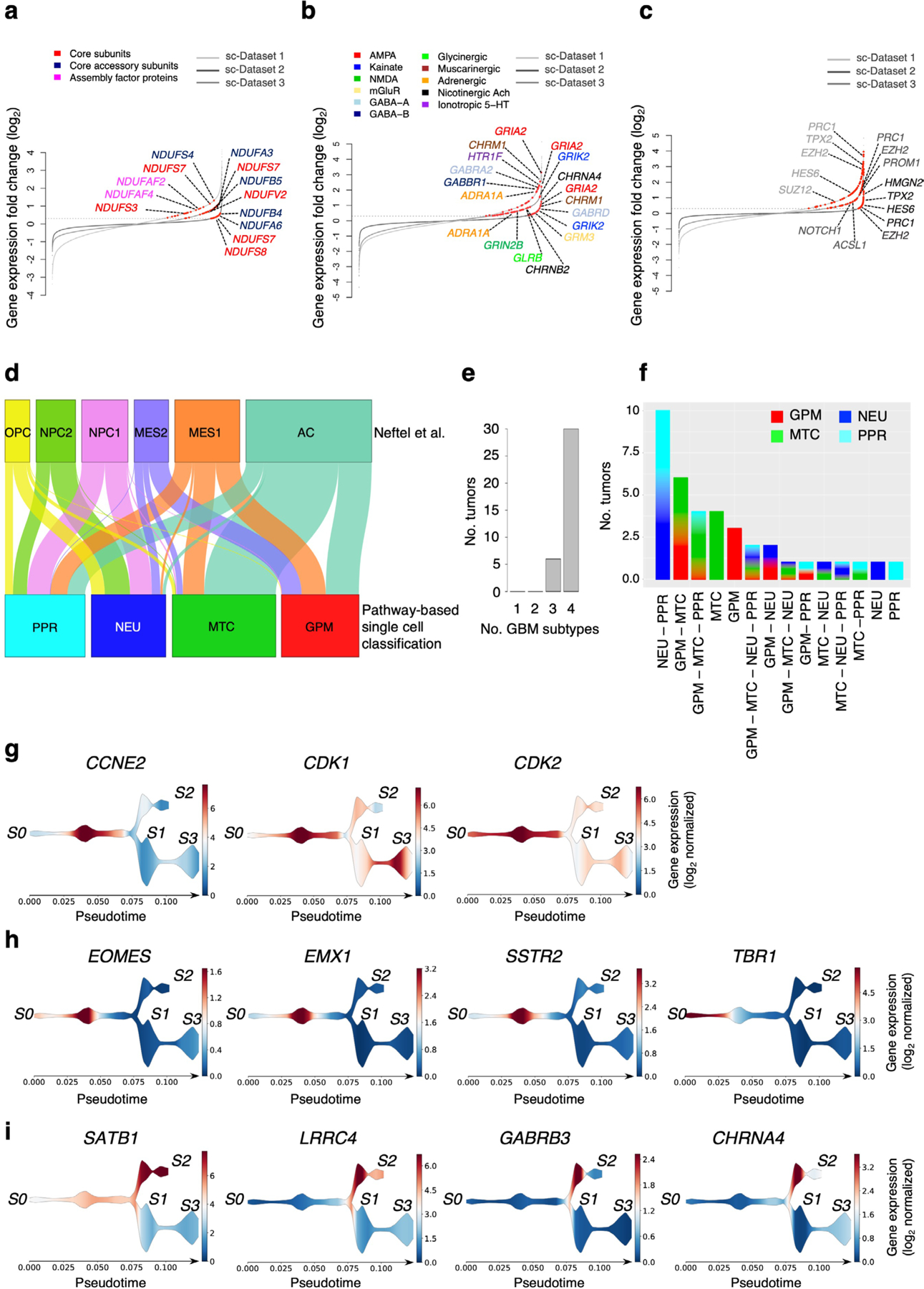
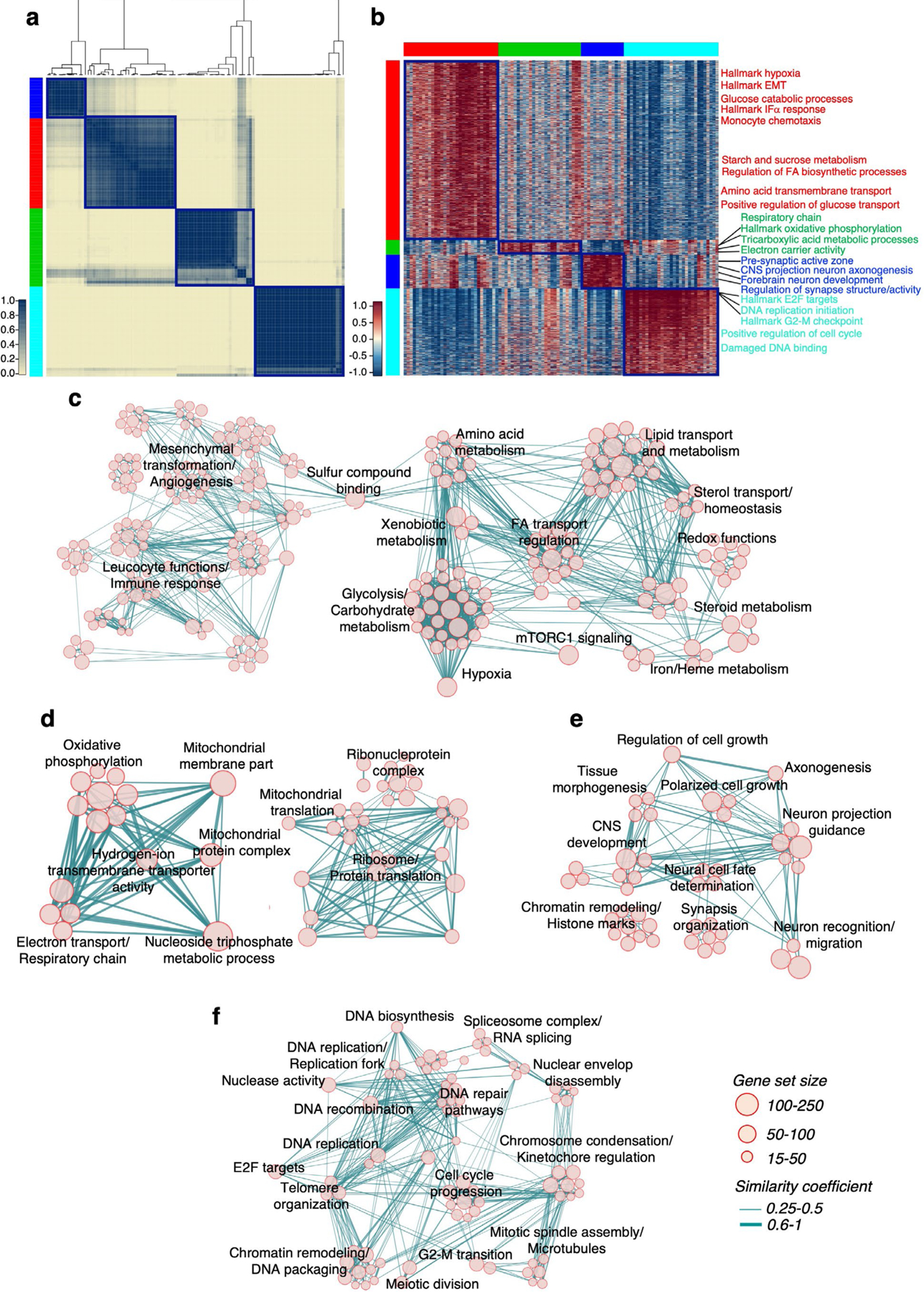
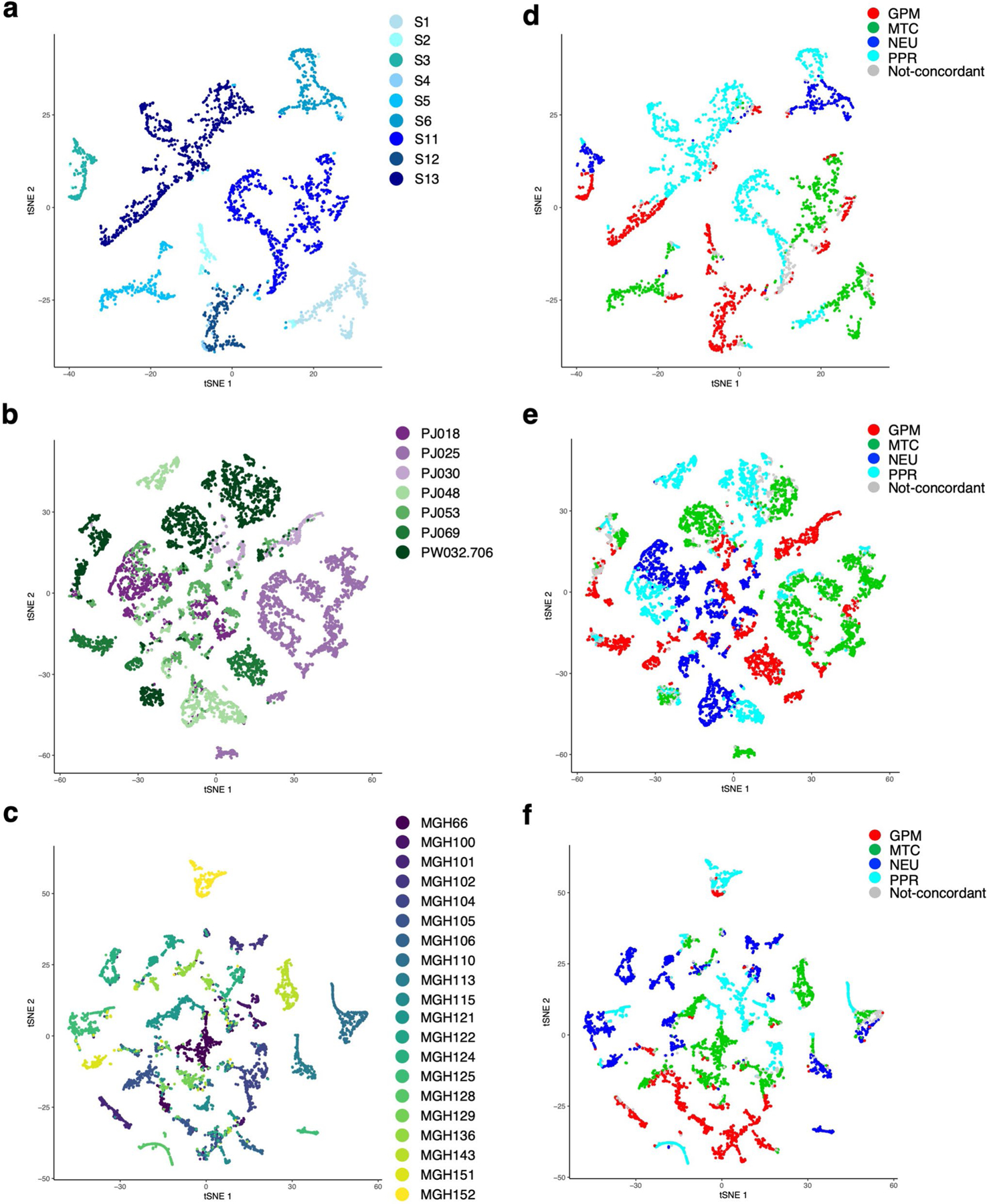
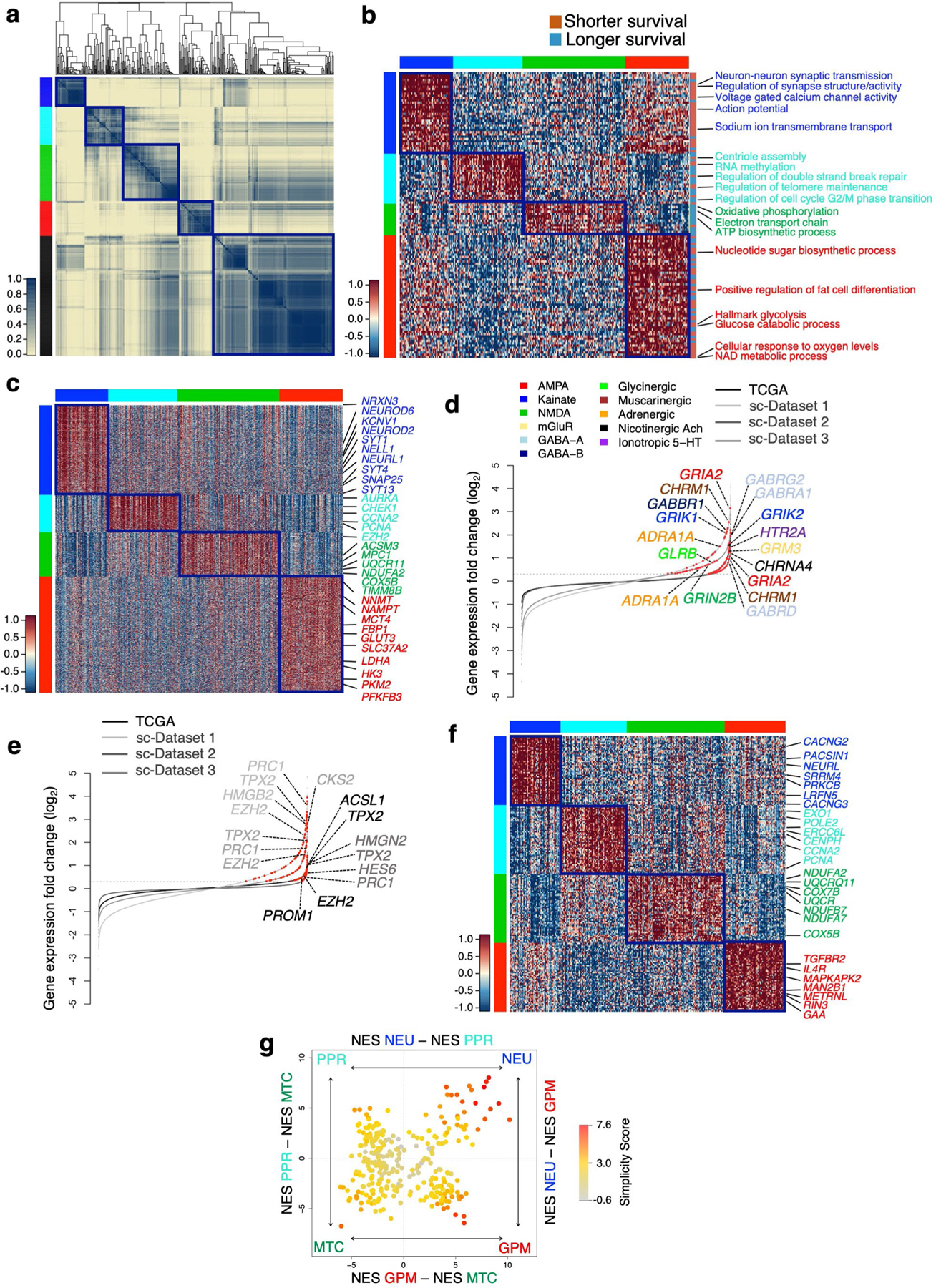
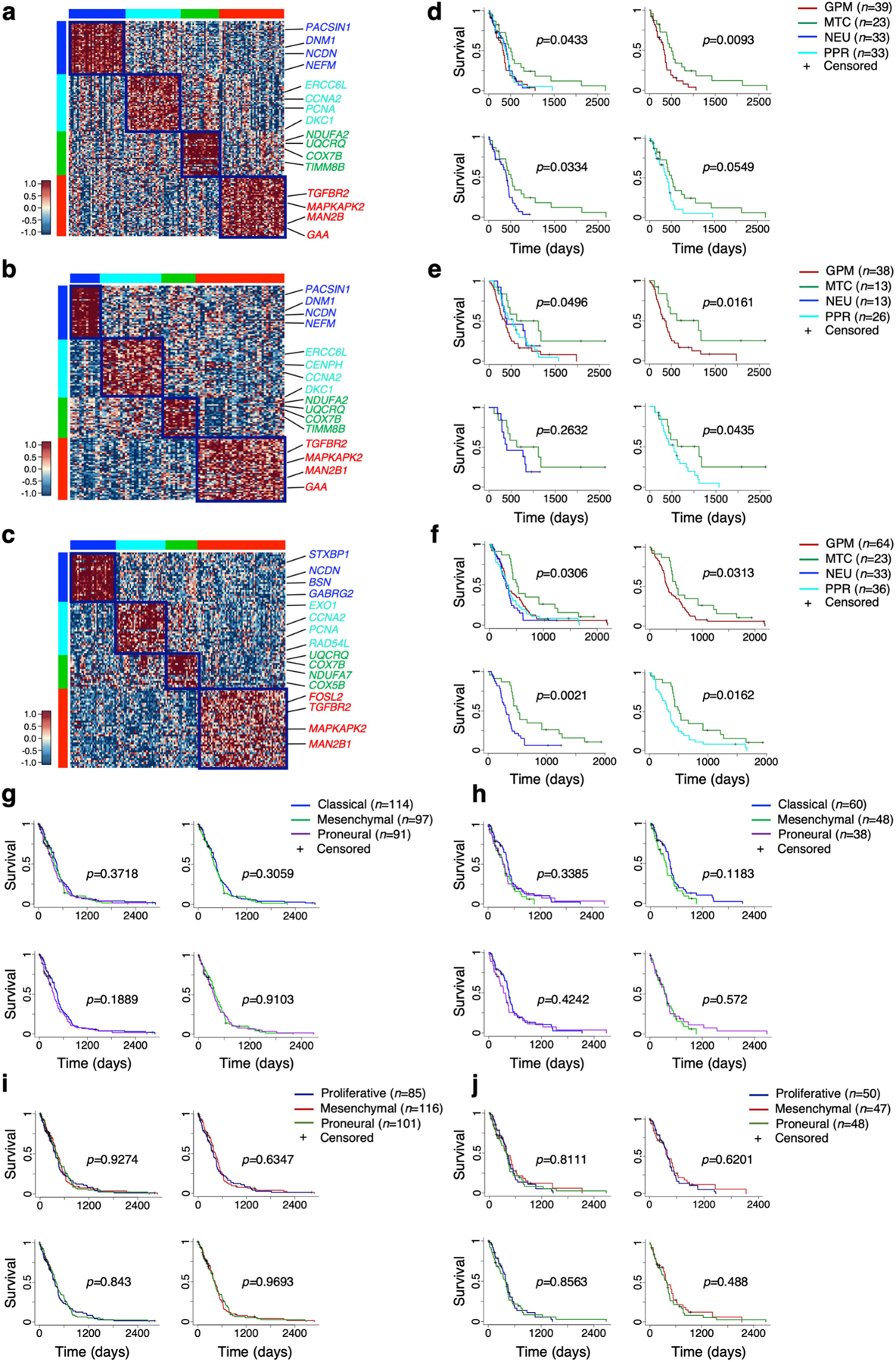
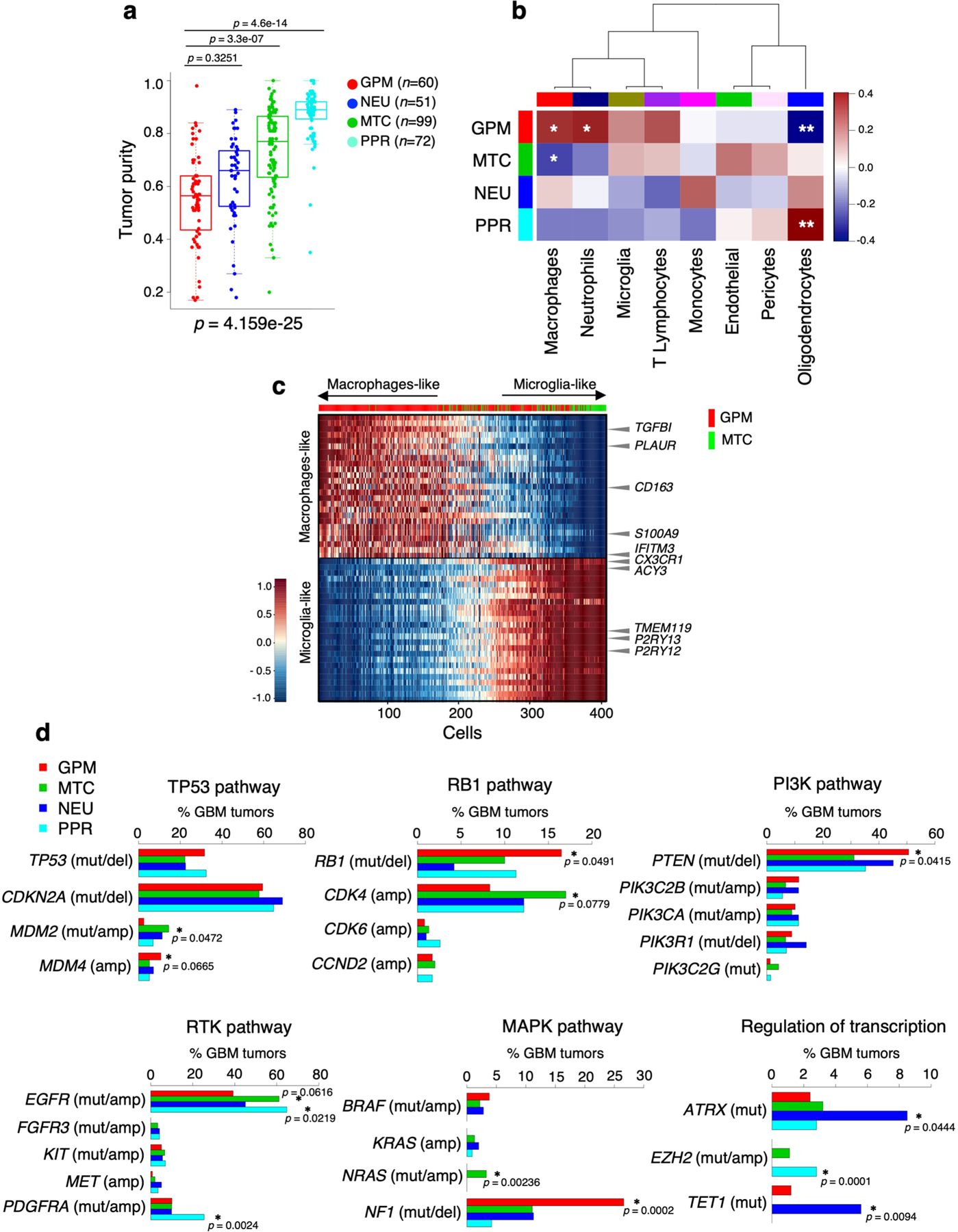
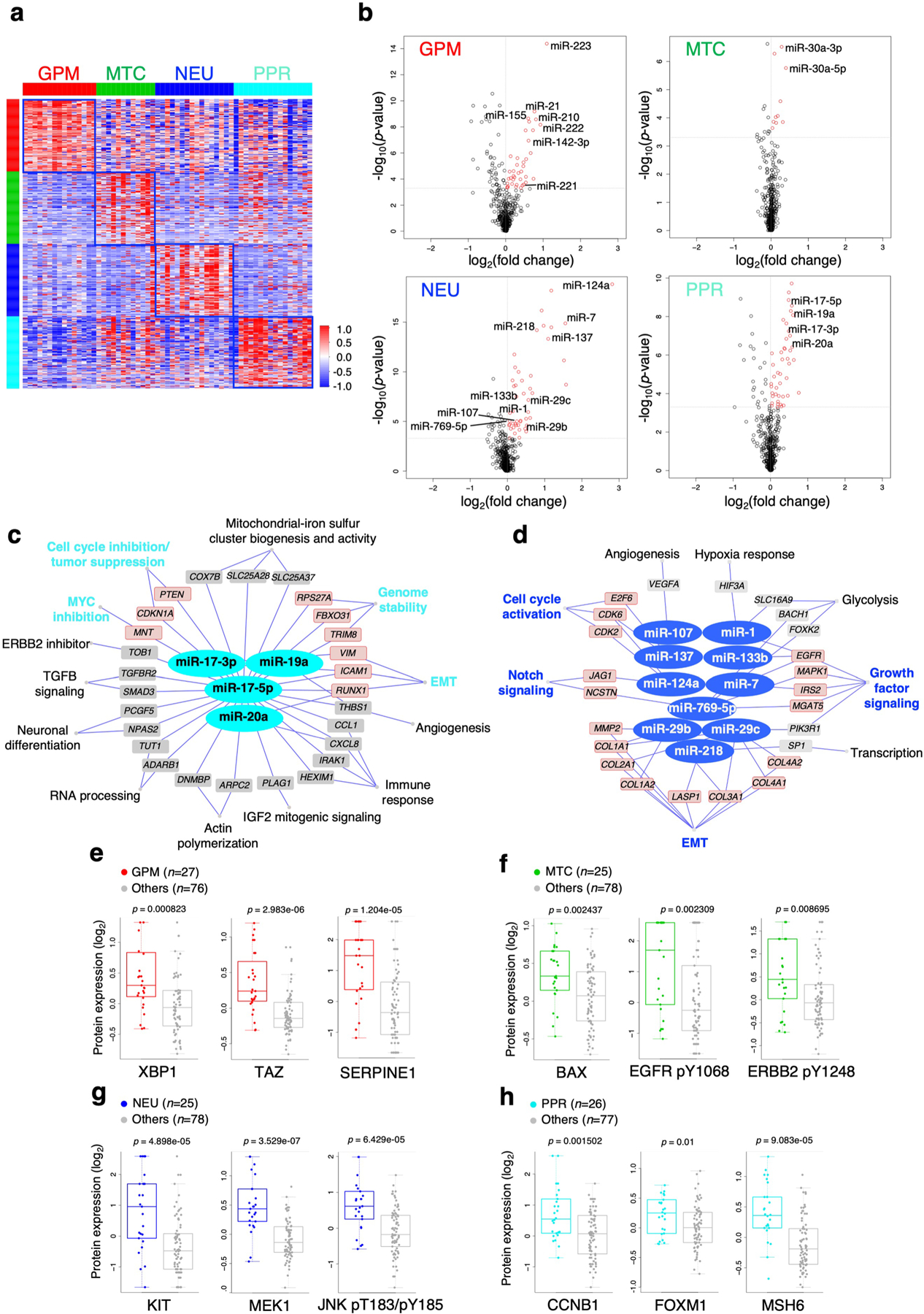
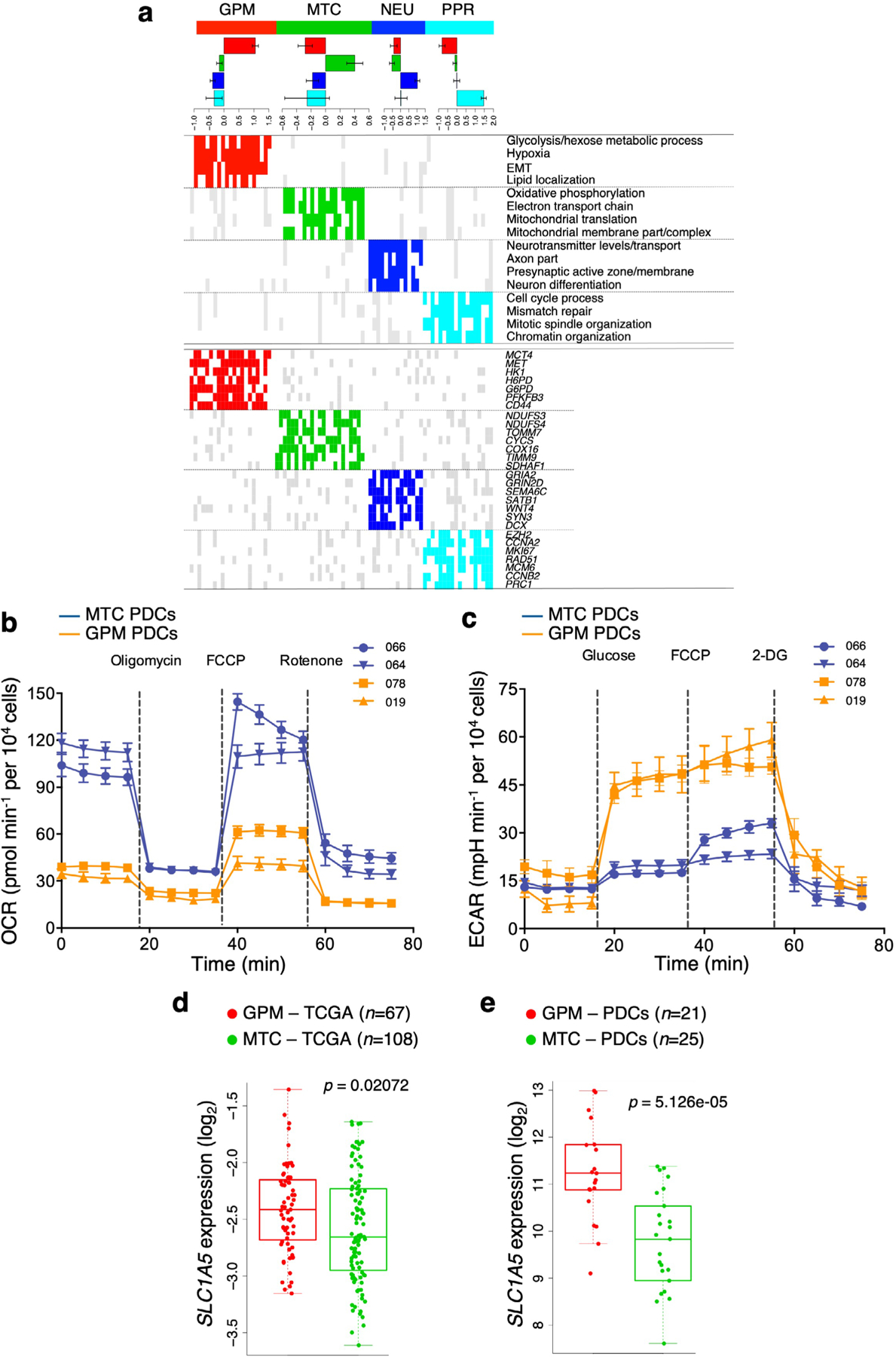
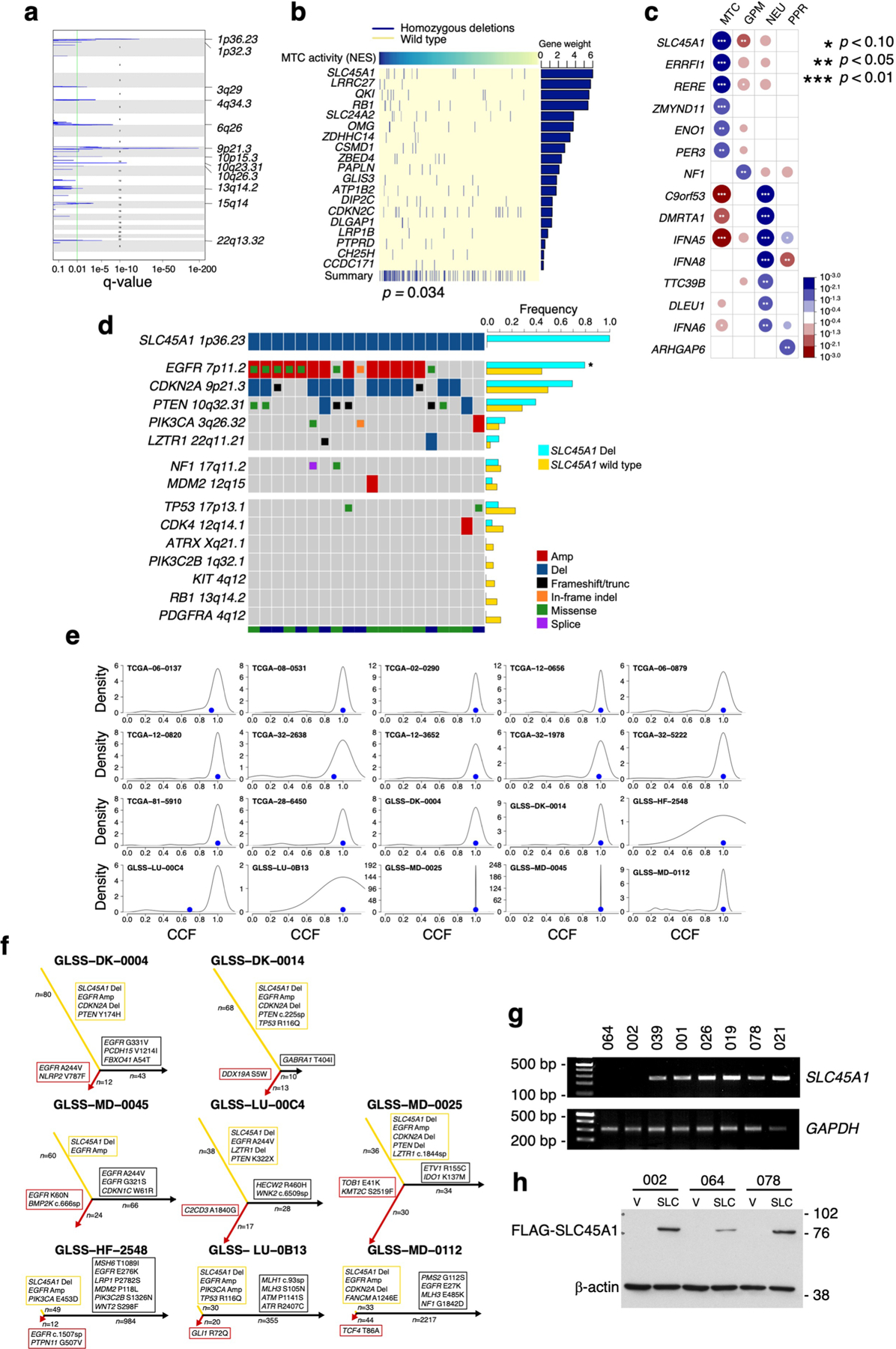
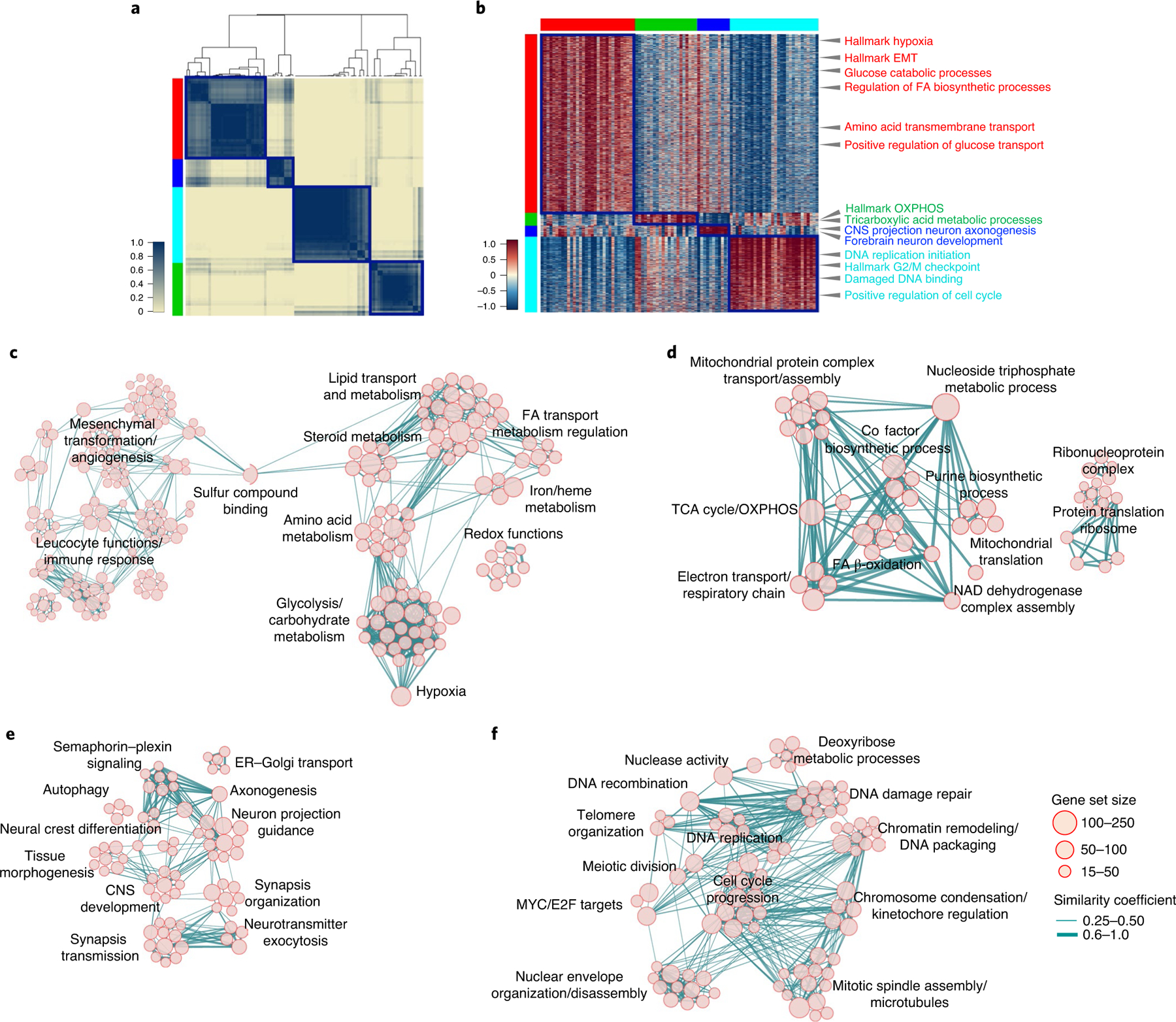
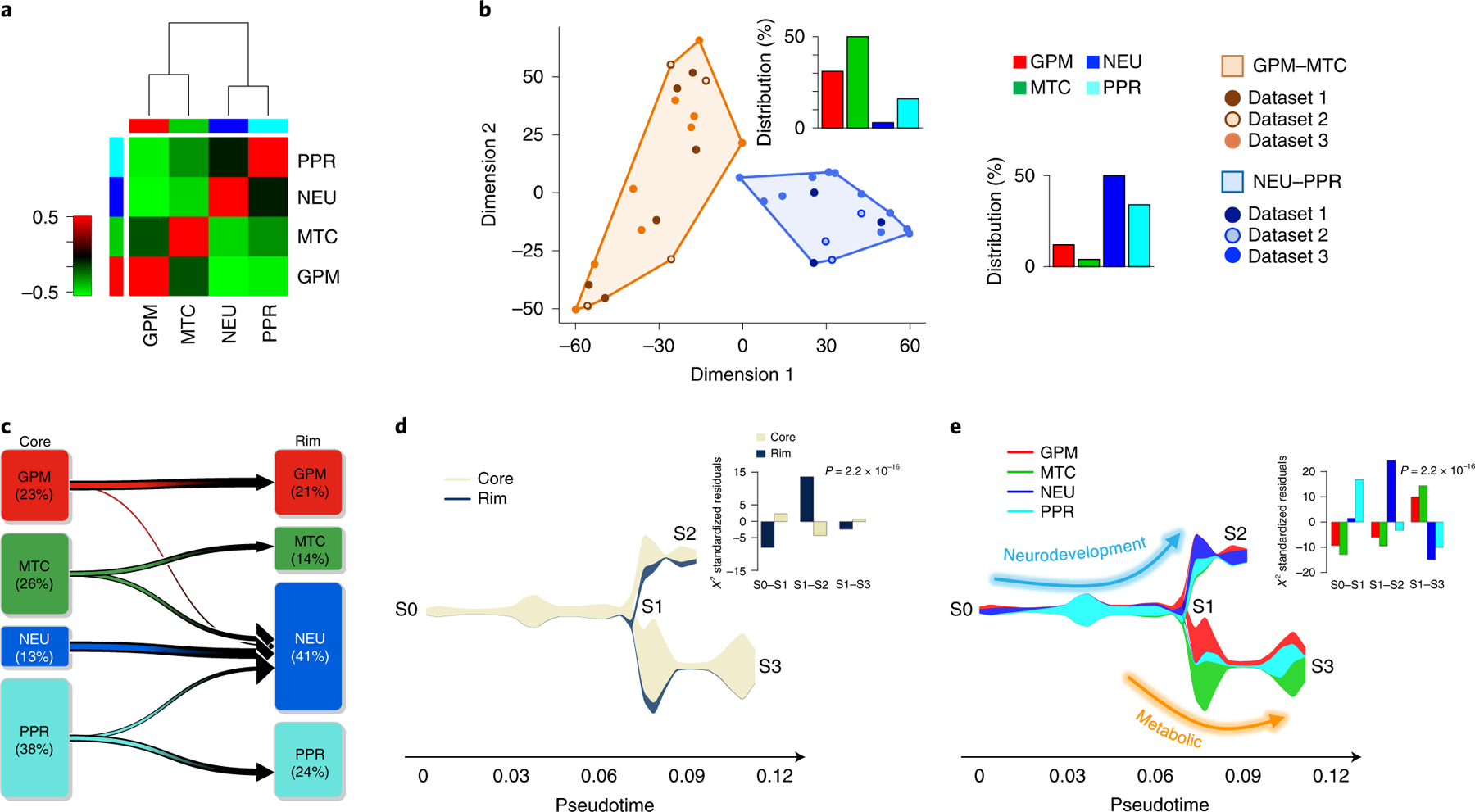
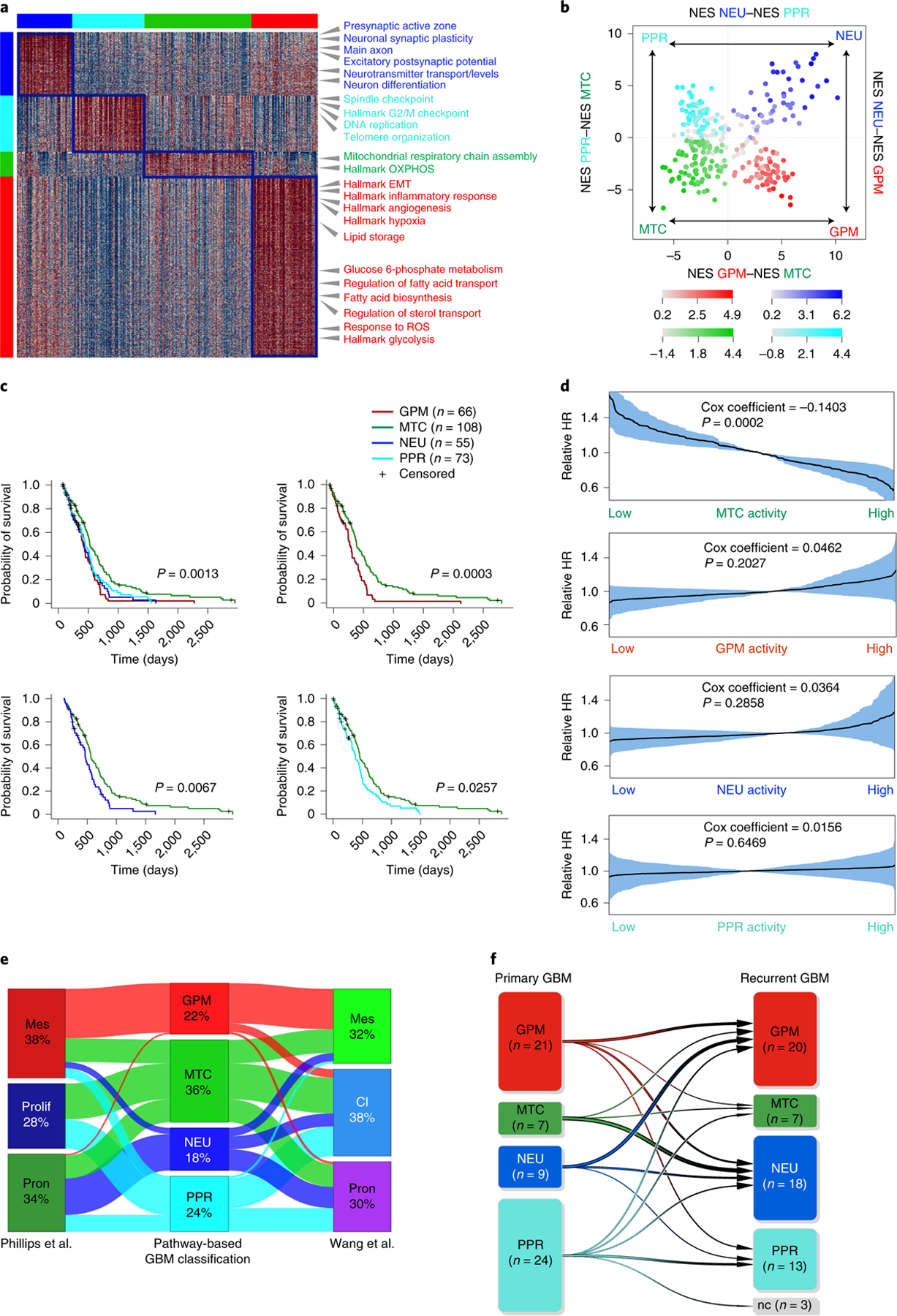
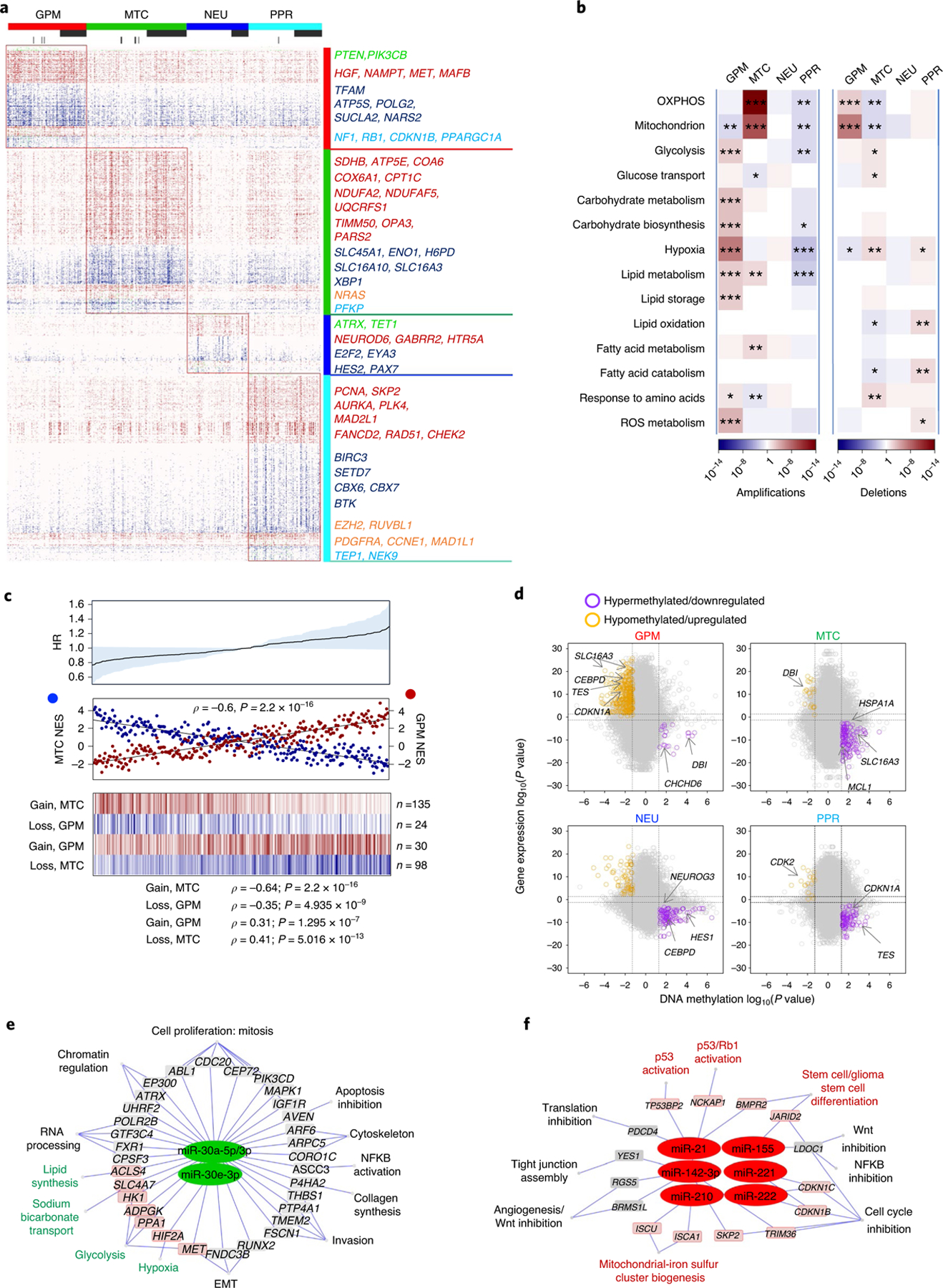
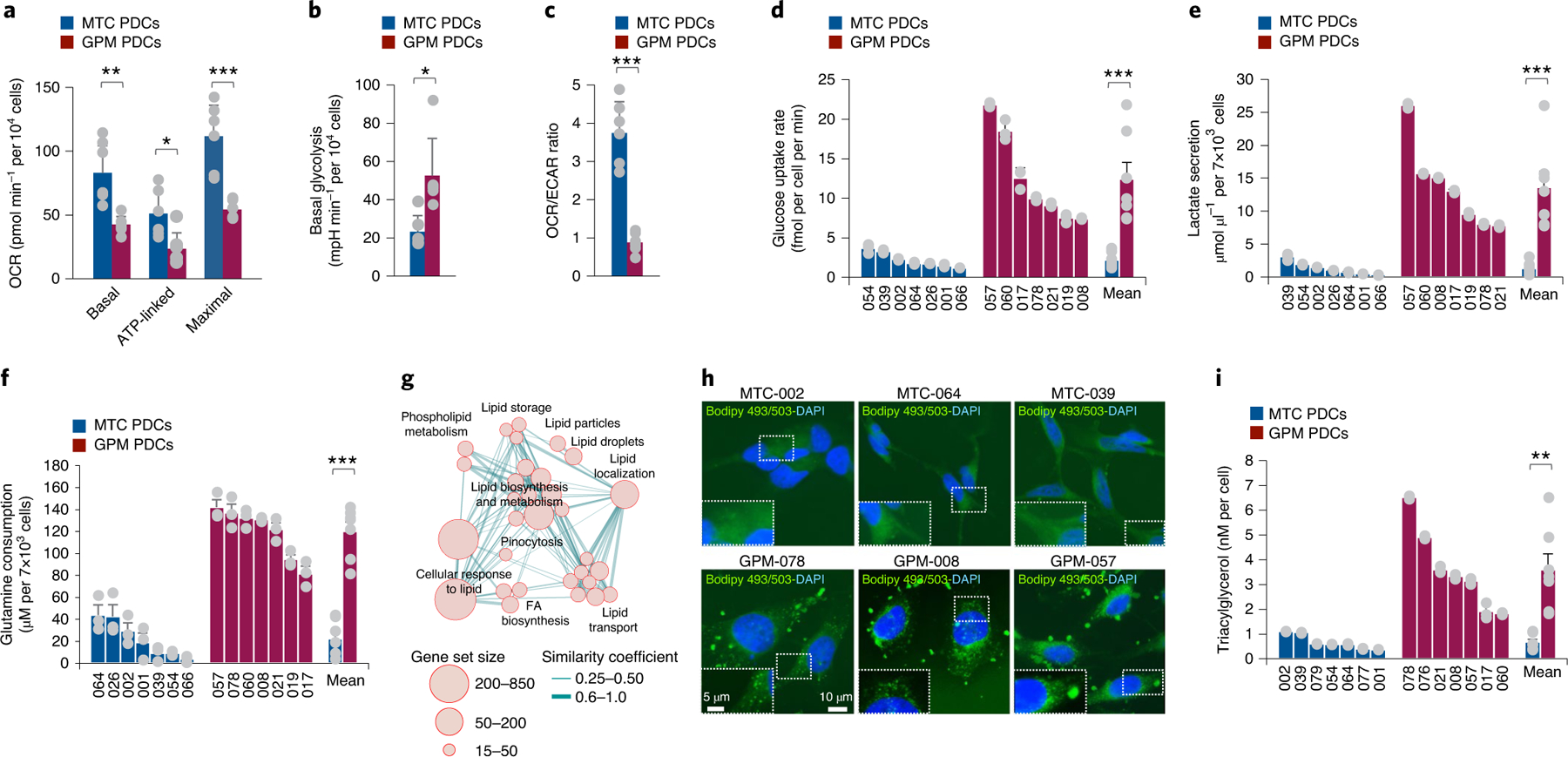
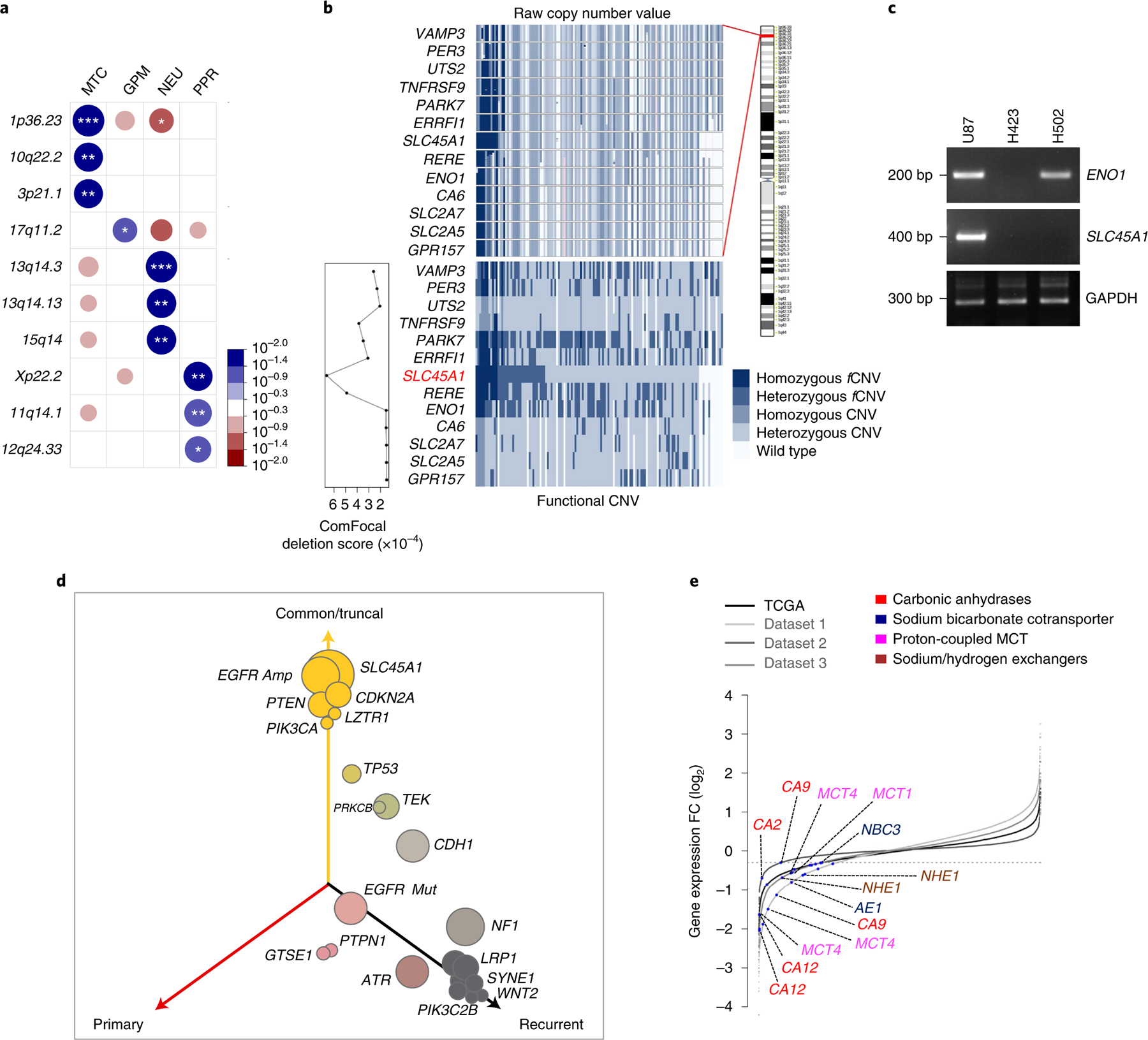
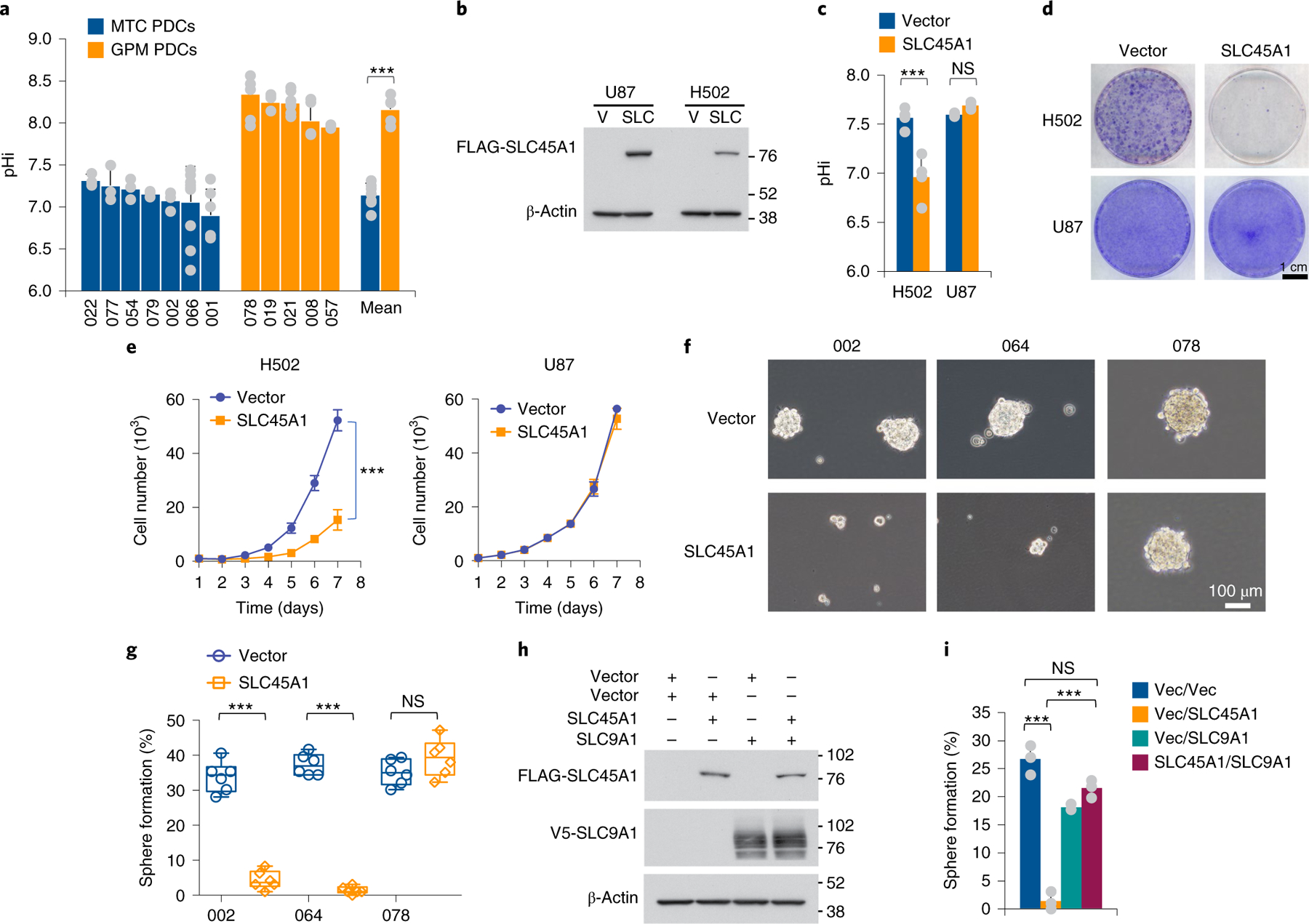
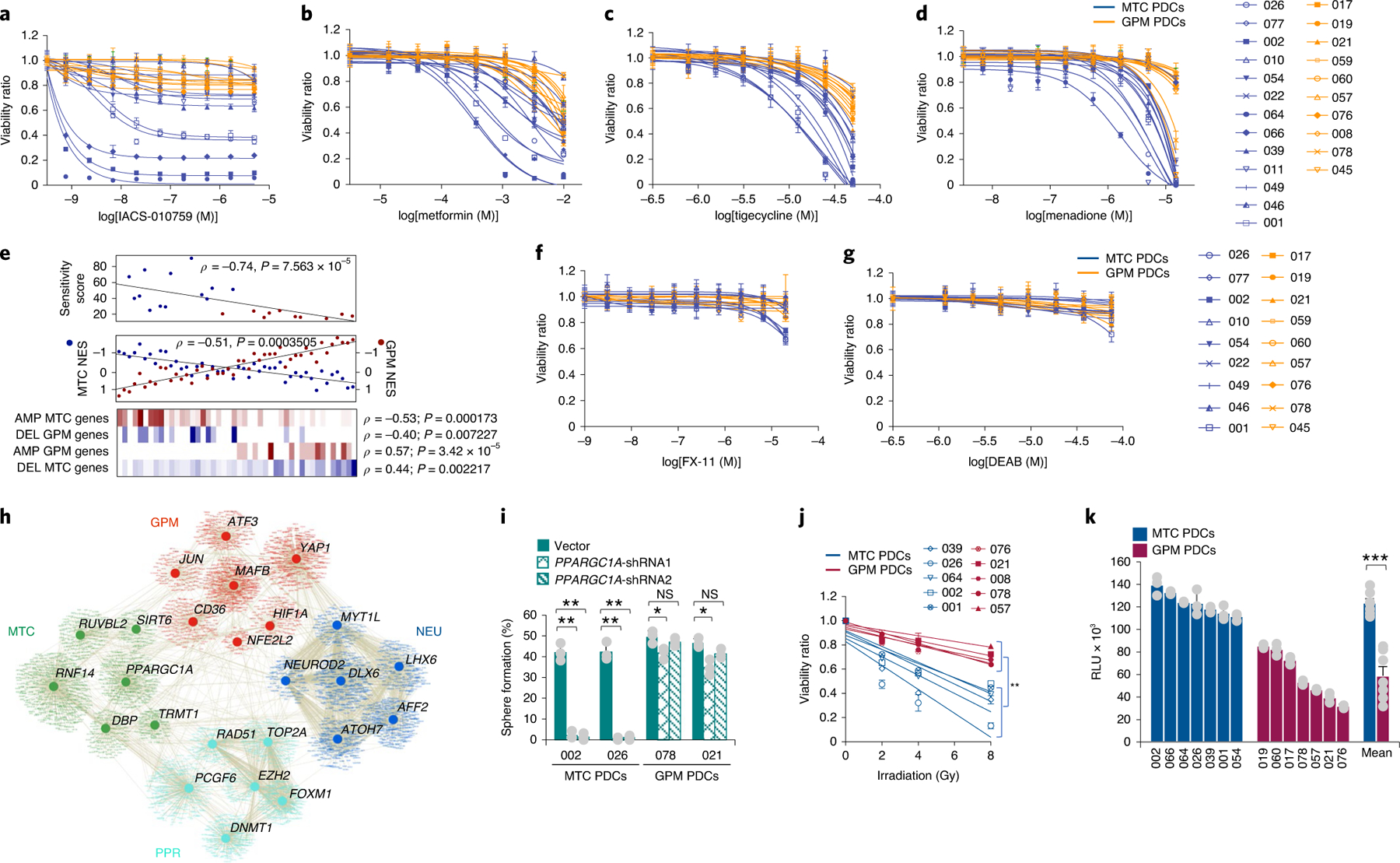
The transcriptomic classification of glioblastoma (GBM) has failed to predict survival and therapeutic vulnerabilities. A computational approach for unbiased identification of core biological traits of single cells and bulk tumors uncovered four tumor cell states and GBM subtypes distributed along neurodevelopmental and metabolic axes, classified as proliferative/progenitor, neuronal, mitochondrial and glycolytic/plurimetabolic. Each subtype was enriched with biologically coherent multiomic features. Mitochondrial GBM was associated with the most favorable clinical outcome. It relied exclusively on oxidative phosphorylation for energy production, whereas the glycolytic/plurimetabolic subtype was sustained by aerobic glycolysis and amino acid and lipid metabolism. Deletion of the glucose-proton symporter SLC45A1 was the truncal alteration most significantly associated with mitochondrial GBM, and the reintroduction of SLC45A1 in mitochondrial glioma cells induced acidification and loss of fitness. Mitochondrial, but not glycolytic/plurimetabolic, GBM exhibited marked vulnerability to inhibitors of oxidative phosphorylation. The pathway-based classification of GBM informs survival and enables precision targeting of cancer metabolism.
Conflict of interest statement
Competing interests A.L. and A.I. are inventors of a biomarker technology that has been licensed to QIAGEN. A.I. received sponsored research funding from AstraZeneca and Taiho Pharmaceutical and has served as a paid consultant/advisor to AIMEDBIO Inc. A.L. received sponsored research funding from Celgene. All other authors declare no competing interests.
Figures
Comment in
-
Pathway-based stratification of glioblastoma.Nat Rev Neurol. 2021 May;17(5):263-264. doi: 10.1038/s41582-021-00474-z. Nat Rev Neurol. 2021. PMID: 33654311 No abstract available.
-
Seeing the GBM diversity spectrum.Nat Cancer. 2021 Feb;2(2):135-137. doi: 10.1038/s43018-021-00176-x. Nat Cancer. 2021. PMID: 35122081 No abstract available.
References
-
- Cieslik M & Chinnaiyan AM Cancer transcriptome profiling at the juncture of clinical translation. Nat. Rev. Genet 19, 93–109 (2018). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
