

Ligand-induced structural changes analysis of ribose-binding protein as studied by molecular dynamics simulations
- PMID: 33682750
- PMCID: PMC8150535
- DOI: 10.3233/THC-218011
Ligand-induced structural changes analysis of ribose-binding protein as studied by molecular dynamics simulations
Abstract
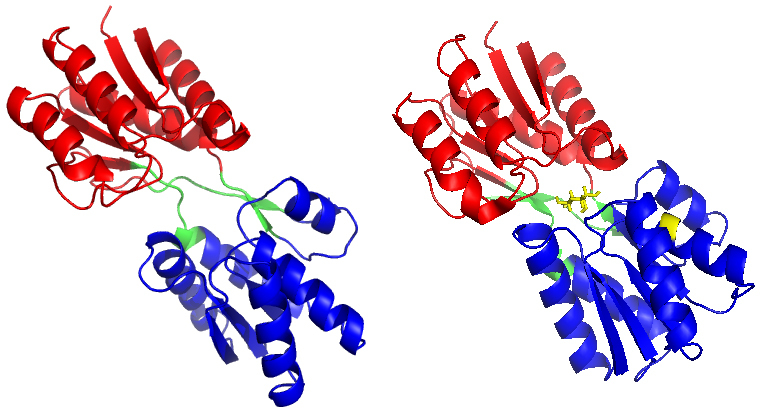
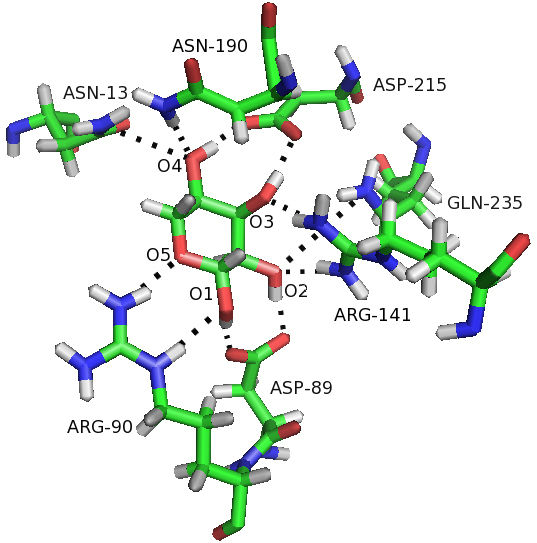
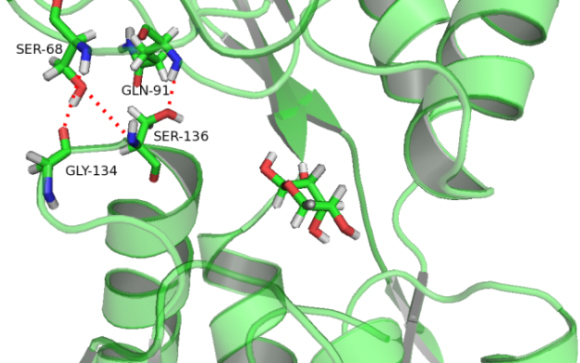
Background: The ribose-binding protein (RBP) from Escherichia coli is one of the representative structures of periplasmic binding proteins. Binding of ribose at the cleft between two domains causes a conformational change corresponding to a closure of two domains around the ligand. The RBP has been crystallized in the open and closed conformations.
Objective: With the complex trajectory as a control, our goal was to study the conformation changes induced by the detachment of the ligand, and the results have been revealed from two computational tools, MD simulations and elastic network models.
Methods: Molecular dynamics (MD) simulations were performed to study the conformation changes of RBP starting from the open-apo, closed-holo and closed-apo conformations.
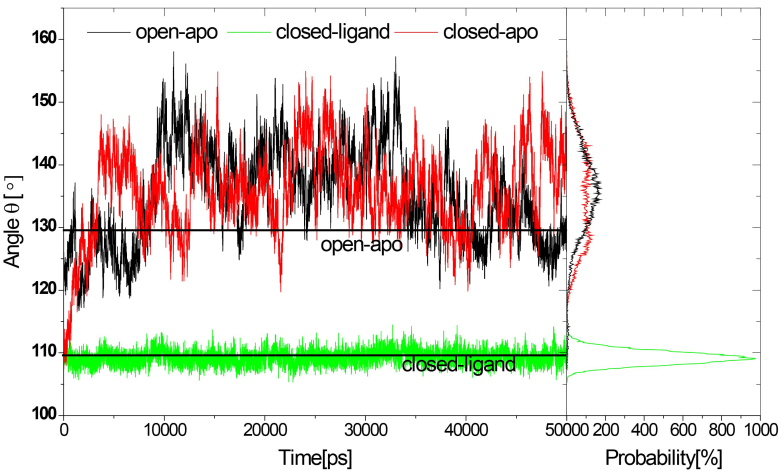
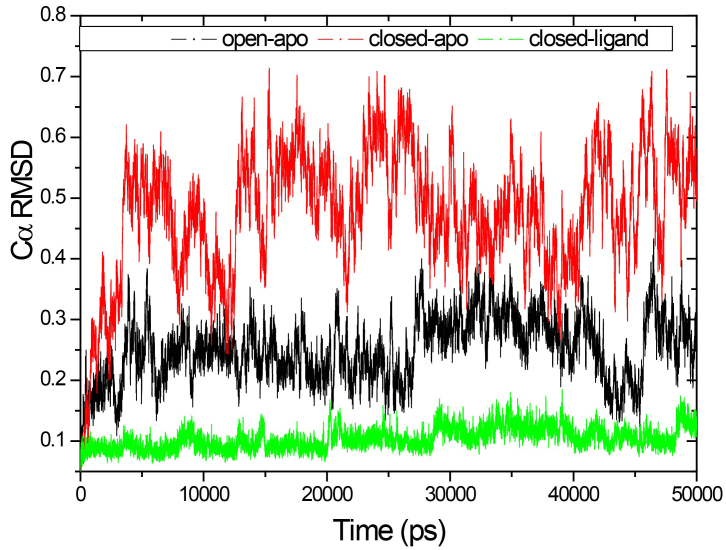
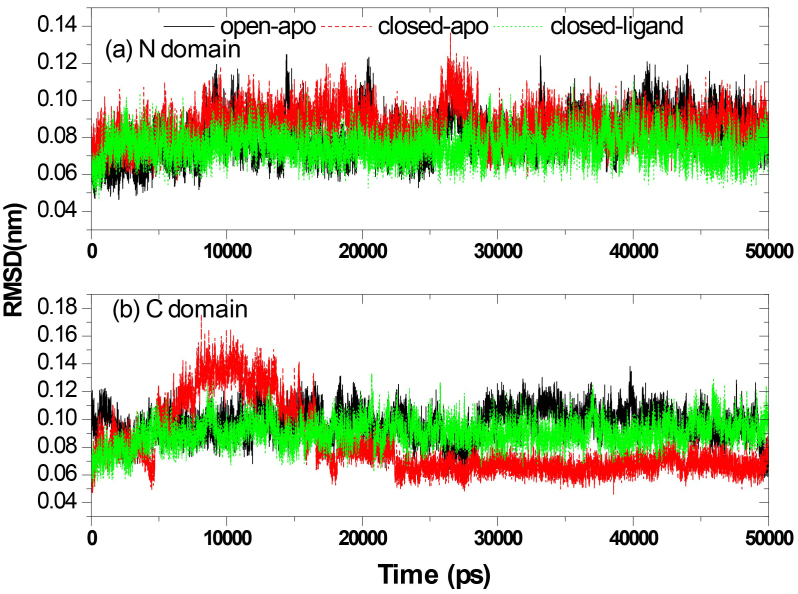
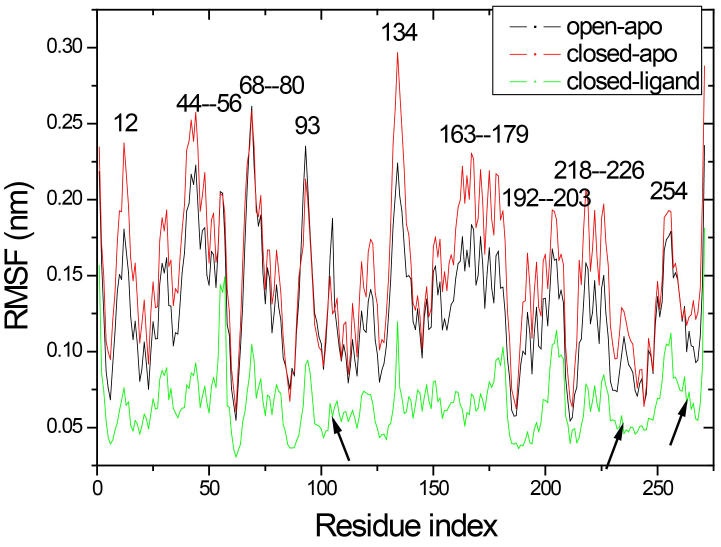
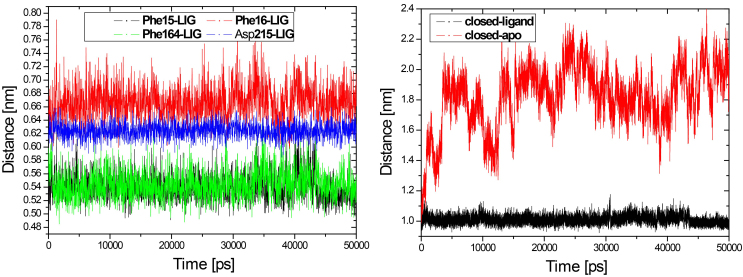
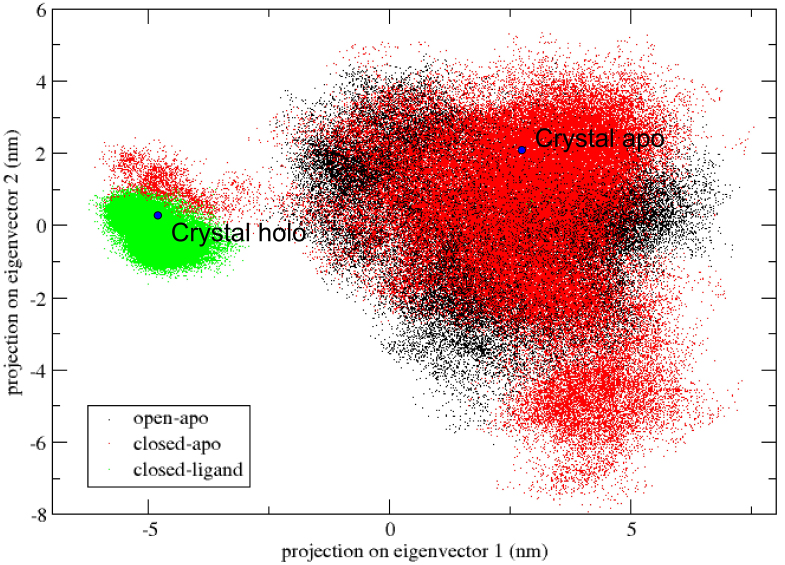
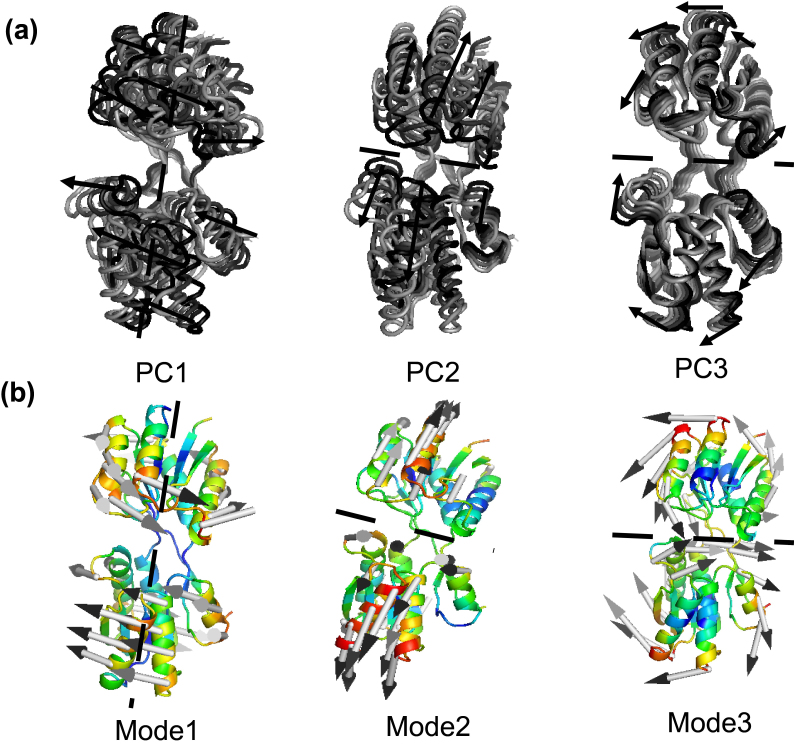
Results: The evolution of the domain opening angle θ clearly indicates large structural changes. The simulations indicate that the closed states in the absence of ribose are inclined to transition to the open states and that ribose-free RBP exists in a wide range of conformations. The first three dominant principal motions derived from the closed-apo trajectories, consisting of rotating, bending and twisting motions, account for the major rearrangement of the domains from the closed to the open conformation.
Conclusions: The motions showed a strong one-to-one correspondence with the slowest modes from our previous study of RBP with the anisotropic network model (ANM). The results obtained for RBP contribute to the generalization of robustness for protein domain motion studies using either the ANM or PCA for trajectories obtained from MD.
Keywords: Ribose-binding protein; conformational change; elastic network model; molecular dynamics simulation.
Conflict of interest statement
None to report.
Figures
Similar articles
-
Analysis of conformational motions and residue fluctuations for Escherichia coli ribose-binding protein revealed with elastic network models.Int J Mol Sci. 2013 May 21;14(5):10552-69. doi: 10.3390/ijms140510552. Int J Mol Sci. 2013. PMID: 23698778 Free PMC article.
-
Unraveling the Coupling between Conformational Changes and Ligand Binding in Ribose Binding Protein Using Multiscale Molecular Dynamics and Free-Energy Calculations.J Phys Chem B. 2021 Mar 25;125(11):2898-2909. doi: 10.1021/acs.jpcb.0c11600. Epub 2021 Mar 17. J Phys Chem B. 2021. PMID: 33728914 Free PMC article.
-
Exploring Large Domain Motions in Proteins Using Atomistic Molecular Dynamics with Enhanced Conformational Sampling.Int J Mol Sci. 2020 Dec 29;22(1):270. doi: 10.3390/ijms22010270. Int J Mol Sci. 2020. PMID: 33383937 Free PMC article.
-
Enzyme dynamics from NMR spectroscopy.Acc Chem Res. 2015 Feb 17;48(2):457-65. doi: 10.1021/ar500340a. Epub 2015 Jan 9. Acc Chem Res. 2015. PMID: 25574774 Free PMC article. Review.
-
A review on description dynamics and conformational changes of proteins using combination of principal component analysis and molecular dynamics simulation.Comput Biol Med. 2024 Dec;183:109245. doi: 10.1016/j.compbiomed.2024.109245. Epub 2024 Oct 9. Comput Biol Med. 2024. PMID: 39388840 Review.
Cited by
-
What have molecular simulations contributed to understanding of Gram-negative bacterial cell envelopes?Microbiology (Reading). 2022 Mar;168(3):001165. doi: 10.1099/mic.0.001165. Microbiology (Reading). 2022. PMID: 35294337 Free PMC article. Review.
-
SPEACH_AF: Sampling protein ensembles and conformational heterogeneity with Alphafold2.PLoS Comput Biol. 2022 Aug 22;18(8):e1010483. doi: 10.1371/journal.pcbi.1010483. eCollection 2022 Aug. PLoS Comput Biol. 2022. PMID: 35994486 Free PMC article.
-
Systematic computational strategies for identifying protein targets and lead discovery.RSC Med Chem. 2024 May 31;15(7):2254-2269. doi: 10.1039/d4md00223g. eCollection 2024 Jul 17. RSC Med Chem. 2024. PMID: 39026640 Free PMC article. Review.
References
-
- Bjorkman AJ, Binnie RA, Zhang H, Cole LB, Hermodson MA, and Mowbray SL. Probing protein-protein interactions. The ribose-binding protein in bacterial transport and chemotaxis. The Journal of Biological Chemistry. 1994; 269: 30206-30211. - PubMed
-
- Bjorkman AJ, and Mowbray SL. Multiple open forms of ribose-binding protein trace the path of its conformational change. Journal of Molecular Biology. 1998; 279: 651-664. - PubMed
-
- Gerstein M, Lesk AM, and Chothia C. Structural mechanisms for domain movements in proteins. Biochemistry. 1994; 33: 6739-6749. - PubMed
-
- Mowbray SL, and Cole LB. 1.7 A X-ray structure of the periplasmic ribose receptor from Escherichia coli. Journal of Molecular Biology. 1992; 225: 155-175. - PubMed
-
- Anderson C, Zucker F, and Steitz T. Space-filling models of kinase clefts and conformation changes. Science. 1979; 204: 375-380. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
