

Clinical Findings and Diagnostic Yield of Arrhythmogenic Cardiomyopathy Through Genomic Screening of Pathogenic or Likely Pathogenic Desmosome Gene Variants
- PMID: 33684294
- PMCID: PMC8284375
- DOI: 10.1161/CIRCGEN.120.003302
Clinical Findings and Diagnostic Yield of Arrhythmogenic Cardiomyopathy Through Genomic Screening of Pathogenic or Likely Pathogenic Desmosome Gene Variants
Abstract
Background: Genomic screening holds great promise for presymptomatic identification of hidden disease, and prevention of dramatic events, including sudden cardiac death associated with arrhythmogenic cardiomyopathy (ACM). Herein, we present findings from clinical follow-up of carriers of ACM-associated pathogenic/likely pathogenic desmosome variants ascertained through genomic screening.
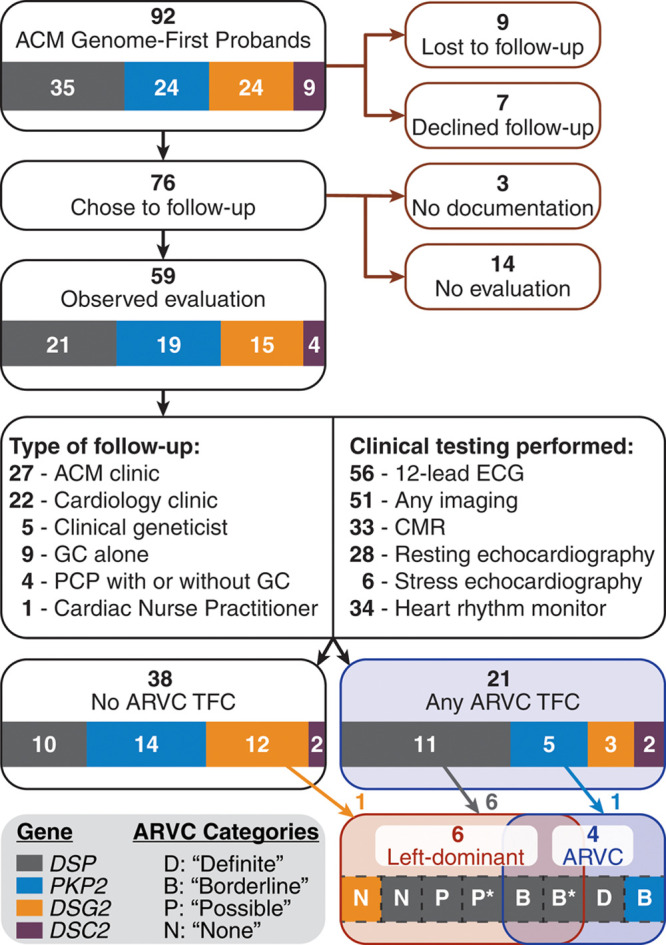
Methods: Of 64 548 eligible participants in Geisinger MyCode Genomic Screening and Counseling program (2015-present), 92 individuals (0.14%) identified with pathogenic/likely pathogenic desmosome variants by clinical laboratory testing were referred for evaluation. We reviewed preresult medical history, patient-reported family history, and diagnostic testing results to assess both arrhythmogenic right ventricular cardiomyopathy and left-dominant ACM.
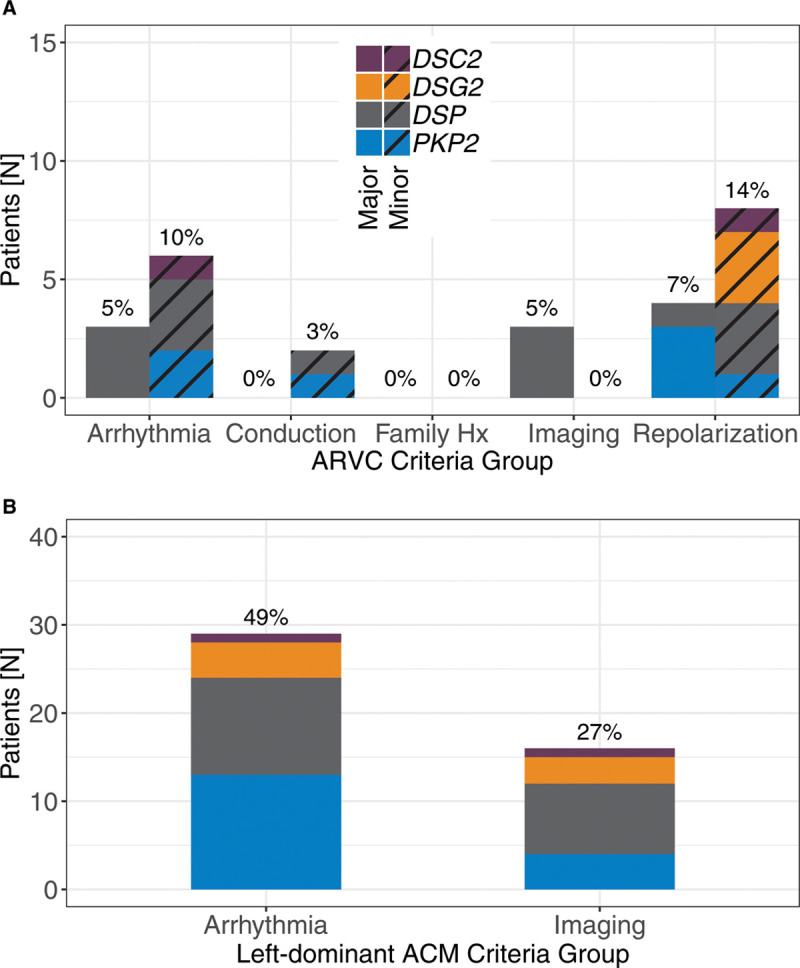
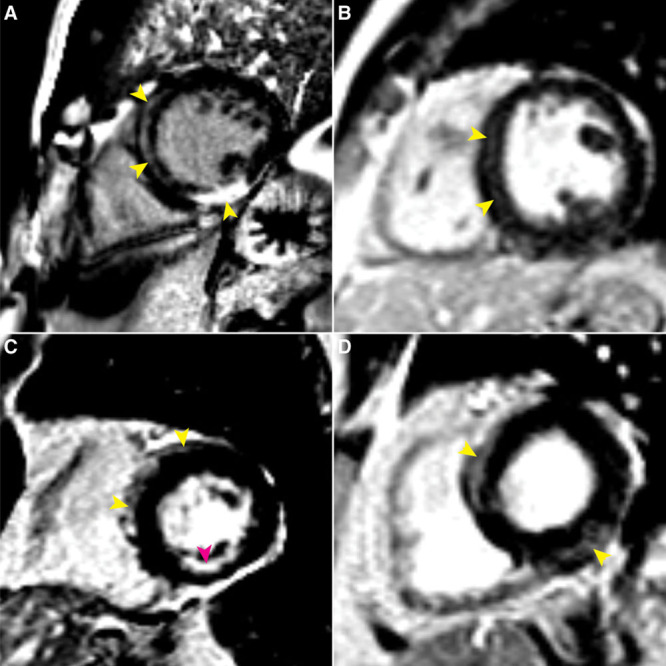
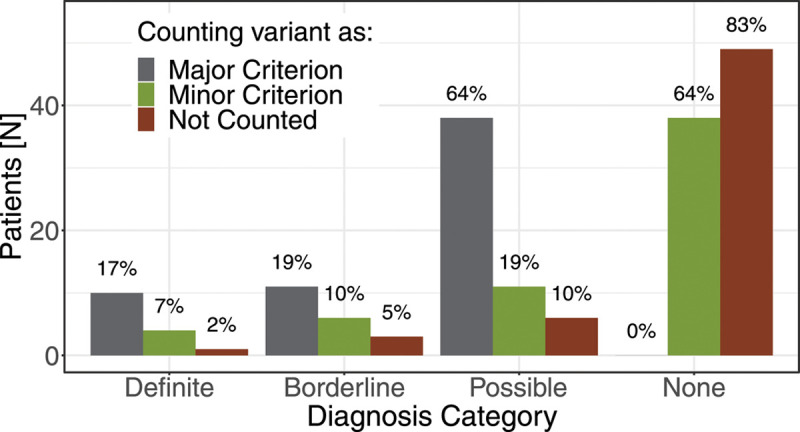
Results: One carrier had a prior diagnosis of dilated cardiomyopathy with arrhythmia; no other related diagnoses or diagnostic family history criteria were reported. Fifty-nine carriers (64%) had diagnostic testing in follow-up. Excluding the variant, 21/59 carriers satisfied at least one arrhythmogenic right ventricular cardiomyopathy task force criterion, 11 (52%) of whom harbored DSP variants, but only 5 exhibited multiple criteria. Six (10%) carriers demonstrated evidence of left-dominant ACM, including high rates of atypical late gadolinium enhancement by magnetic resonance imaging and nonsustained ventricular tachycardia. Two individuals received new cardiomyopathy diagnoses and received defibrillators for primary prevention.
Conclusions: Genomic screening for pathogenic/likely pathogenic variants in desmosome genes can uncover both left- and right-dominant ACM. Findings of overt cardiomyopathy were limited but were most common in DSP-variant carriers and notably absent in PKP2-variant carriers. Consideration of the pathogenic/likely pathogenic variant as a major criterion for diagnosis is inappropriate in the setting of genomic screening.
Keywords: arrhythmogenic right ventricular cardiomyopathy; desmosome; genetic screening; genomics; sudden cardiac death.
Figures
References
-
- Antman EM, Loscalzo J. Precision medicine in cardiology. Nat Rev Cardiol. 2016; 13:591–602. doi: 10.1038/nrcardio.2016.101 - PubMed
-
- Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, Herman GE, Hufnagel SB, Klein TE, Korf BR, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017; 19:249–255. doi: 10.1038/gim.2016.190 - PubMed
-
- Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FCC, Daubert JP, de Chillou C, DePasquale EC, Desai MY, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019; 16:e301–e372. doi: 10.1016/j.hrthm.2019.05.007 - PubMed
-
- Dalal D, Nasir K, Bomma C, Prakasa K, Tandri H, Piccini J, Roguin A, Tichnell C, James C, Russell SD, et al. Arrhythmogenic right ventricular dysplasia: a United States experience. Circulation. 2005; 112:3823–3832. doi: 10.1161/CIRCULATIONAHA.105.542266 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
