

Chemotherapeutic drugs: Cell death- and resistance-related signaling pathways. Are they really as smart as the tumor cells?
- PMID: 33684837
- PMCID: PMC7938256
- DOI: 10.1016/j.tranon.2021.101056
Chemotherapeutic drugs: Cell death- and resistance-related signaling pathways. Are they really as smart as the tumor cells?
Abstract
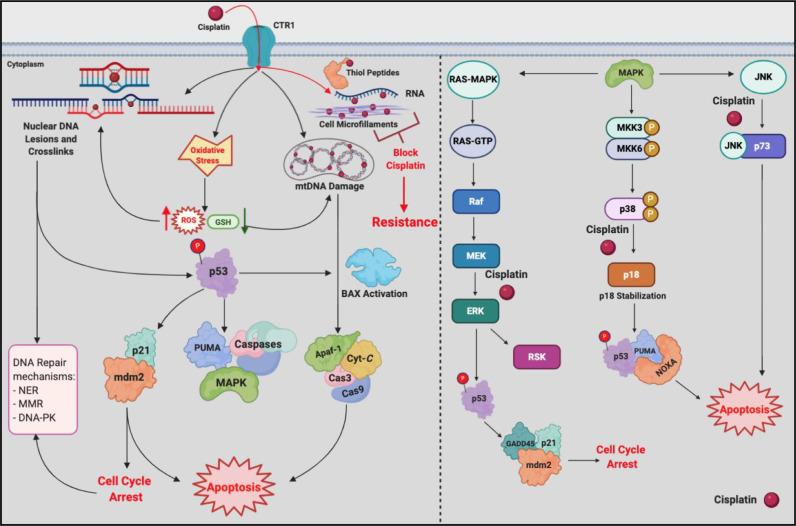
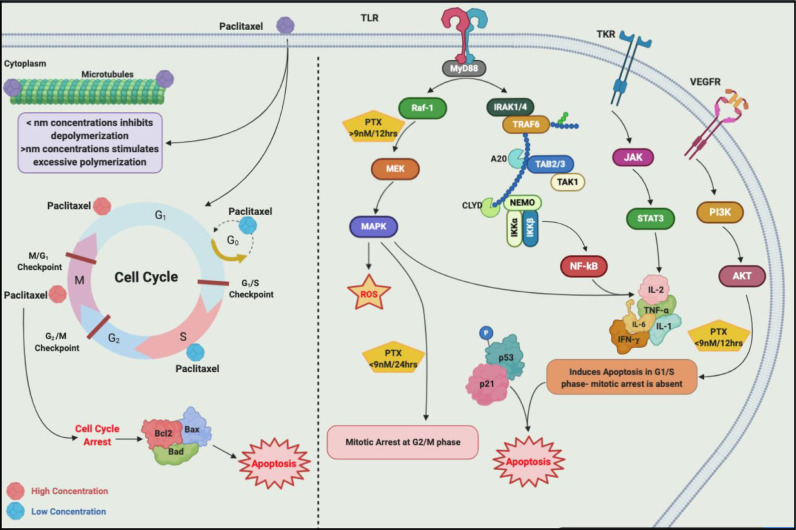
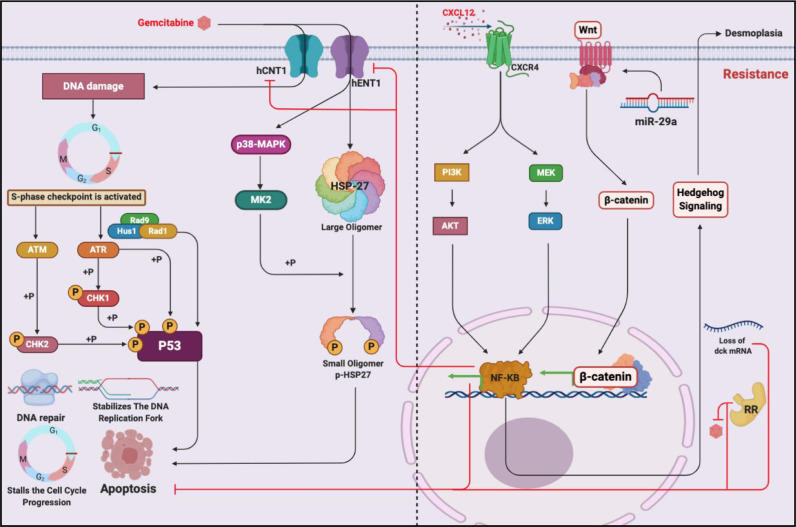
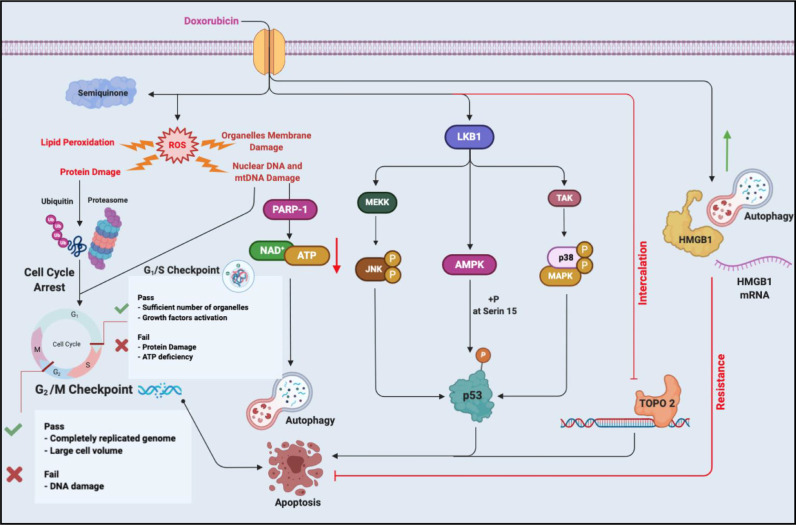
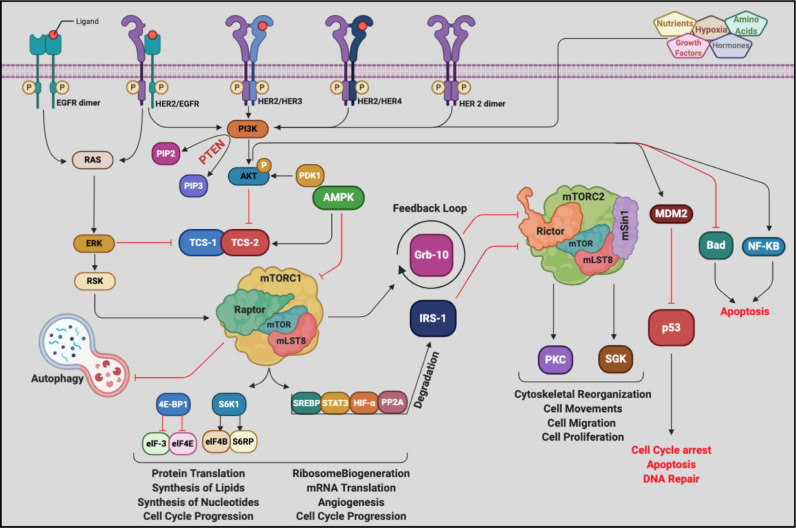
Chemotherapeutic drugs kill cancer cells or control their progression all over the patient's body, while radiation- and surgery-based treatments perform in a particular site. Based on their mechanisms of action, they are classified into different groups, including alkylating substrates, antimetabolite agents, anti-tumor antibiotics, inhibitors of topoisomerase I and II, mitotic inhibitors, and finally, corticosteroids. Although chemotherapeutic drugs have brought about more life expectancy, two major and severe complications during chemotherapy are chemoresistance and tumor relapse. Therefore, we aimed to review the underlying intracellular signaling pathways involved in cell death and resistance in different chemotherapeutic drug families to clarify the shortcomings in the conventional single chemotherapy applications. Moreover, we have summarized the current combination chemotherapy applications, including numerous combined-, and encapsulated-combined-chemotherapeutic drugs. We further discussed the possibilities and applications of precision medicine, machine learning, next-generation sequencing (NGS), and whole-exome sequencing (WES) in promoting cancer immunotherapies. Finally, some of the recent clinical trials concerning the application of immunotherapies and combination chemotherapies were included as well, in order to provide a practical perspective toward the future of therapies in cancer cases.
Keywords: Chemoresistance; Chemotherapeutic drugs; Combination chemotherapy; Death-related intracellular signaling; Intracellular signaling; Precision medicine; Resistance-related intracellular signaling.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Competing Interest The authors declare no conflict of interest.
Figures



References
-
- Raimondi S., Maisonneuve P., Lowenfels A.B. Epidemiology of pancreatic cancer: an overview. Nat. Rev. Gastroenterol. Hepatol. 2009;6:699. - PubMed
-
- Midha S., Chawla S., Garg P.K. Modifiable and non-modifiable risk factors for pancreatic cancer: a review. Cancer Lett. 2016;381:269–277. - PubMed
-
- Kushi L.H., Doyle C., McCullough M., Rock C.L., Demark-Wahnefried W., Bandera E.V., Gapstur S., Patel A.V., Andrews K., Gansler T. American cancer society guidelines on nutrition and physical activity for cancer prevention: reducing the risk of cancer with healthy food choices and physical activity. CA: Cancer J. Clin. 2012;62:30–67. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
