

Organic dust-induced mitochondrial dysfunction could be targeted via cGAS-STING or cytoplasmic NOX-2 inhibition using microglial cells and brain slice culture models
- PMID: 33687557
- PMCID: PMC8154696
- DOI: 10.1007/s00441-021-03422-x
Organic dust-induced mitochondrial dysfunction could be targeted via cGAS-STING or cytoplasmic NOX-2 inhibition using microglial cells and brain slice culture models
Abstract
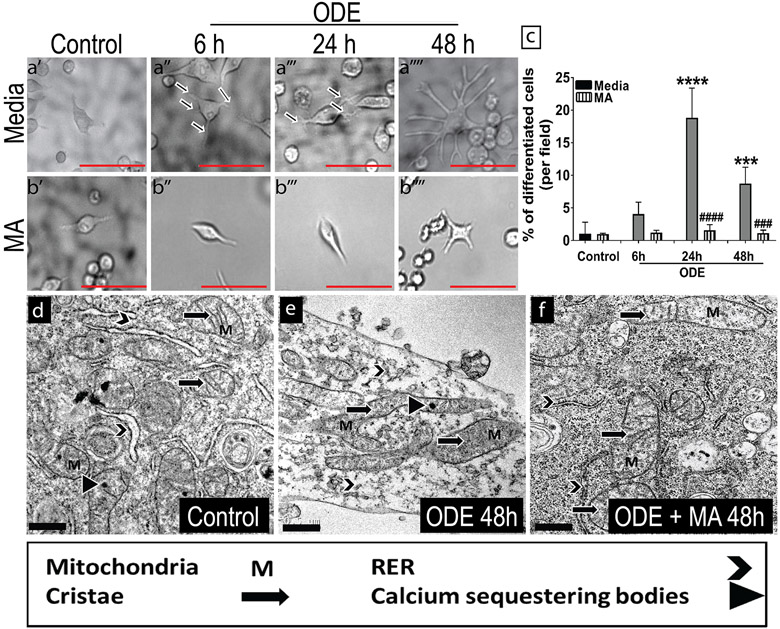
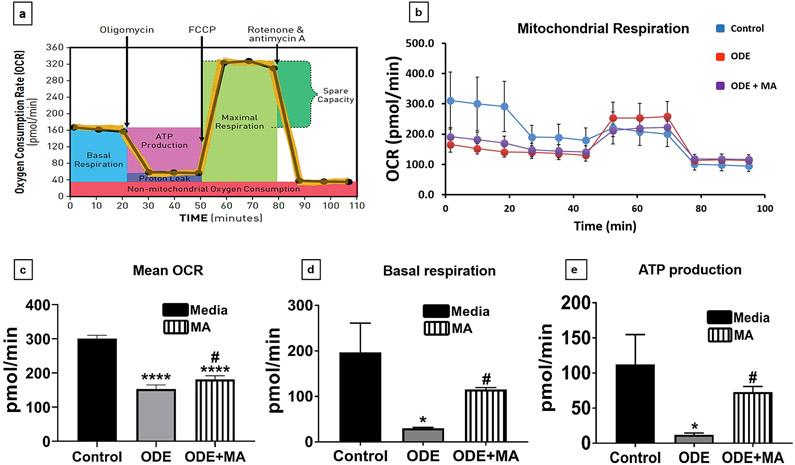
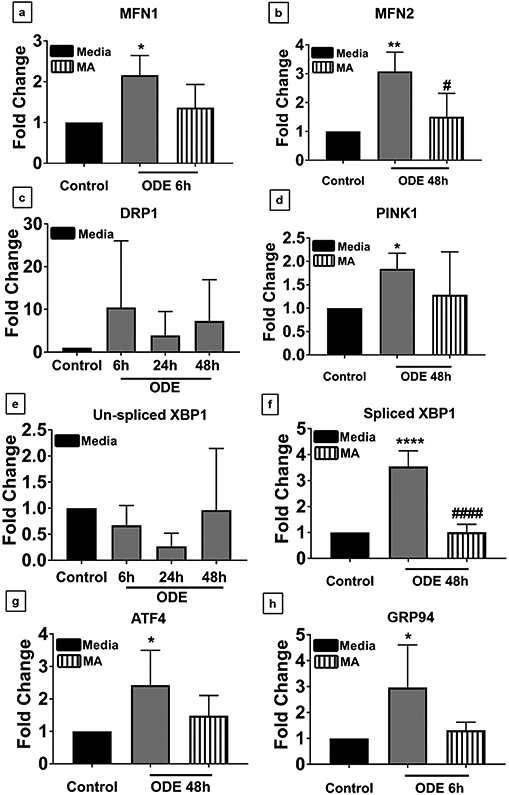
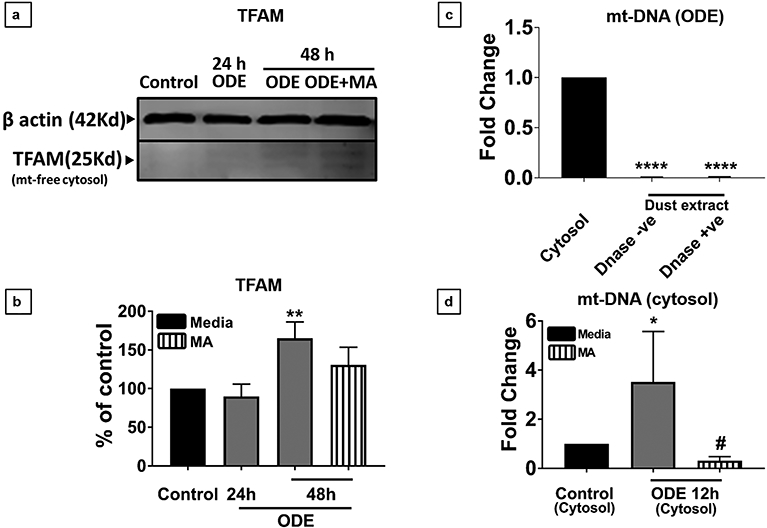
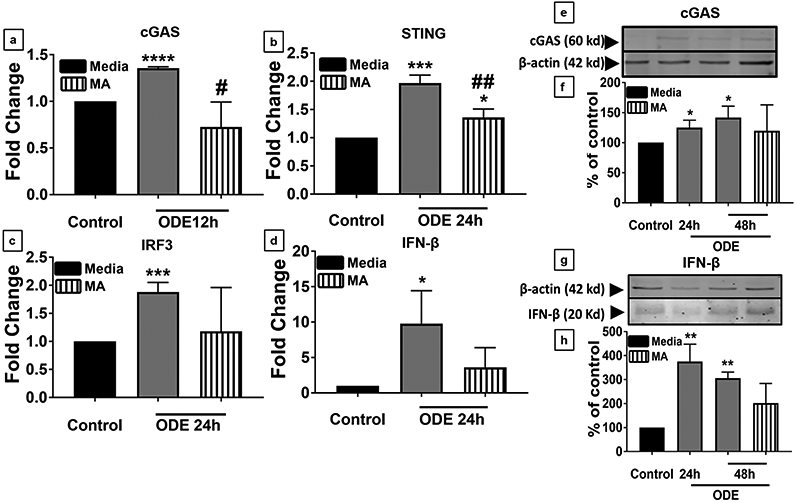
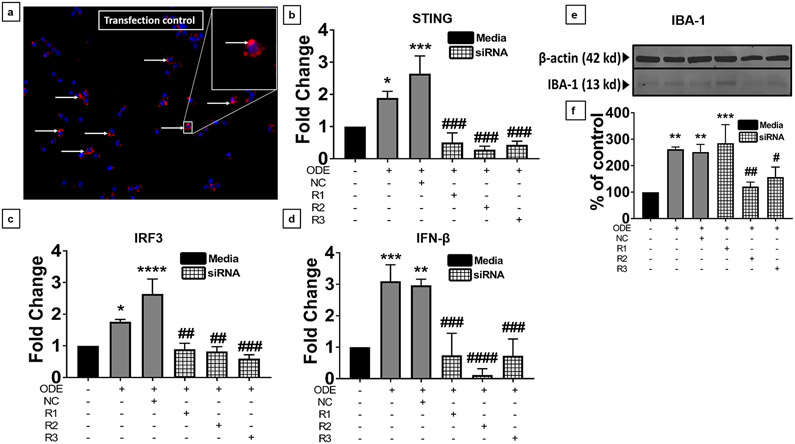
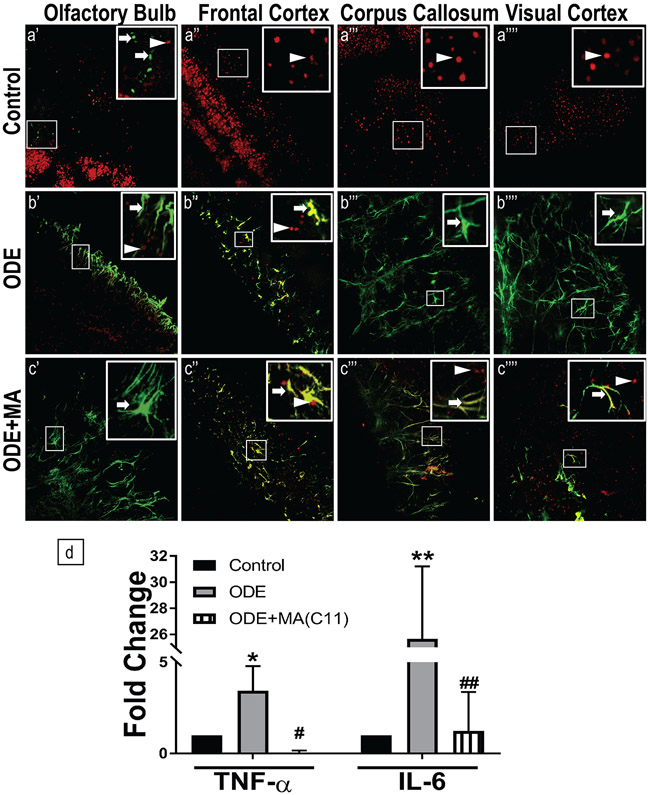
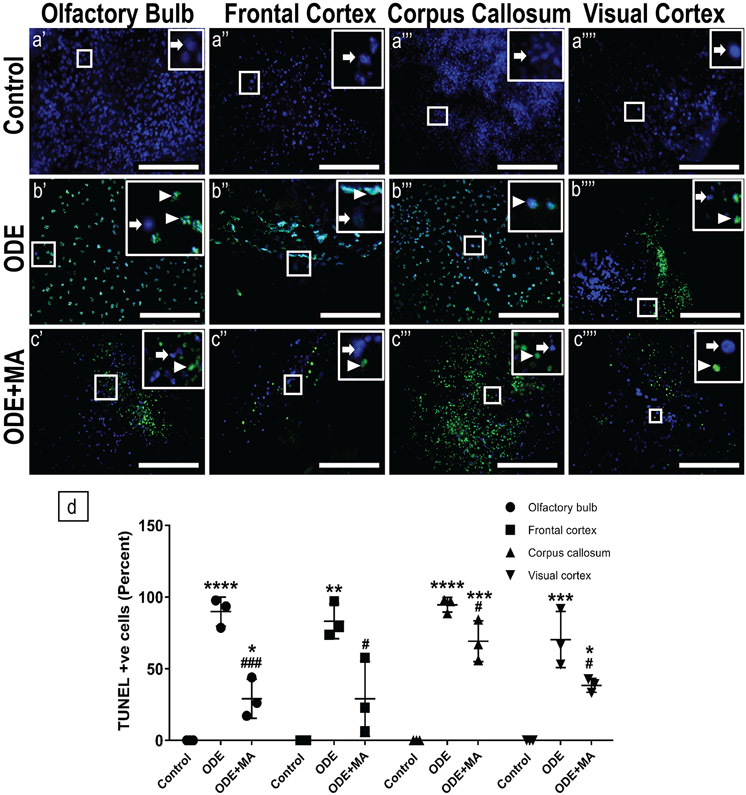
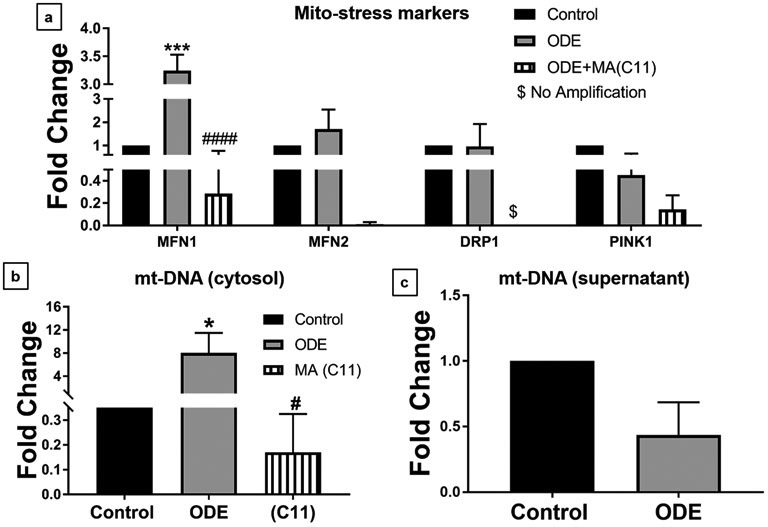
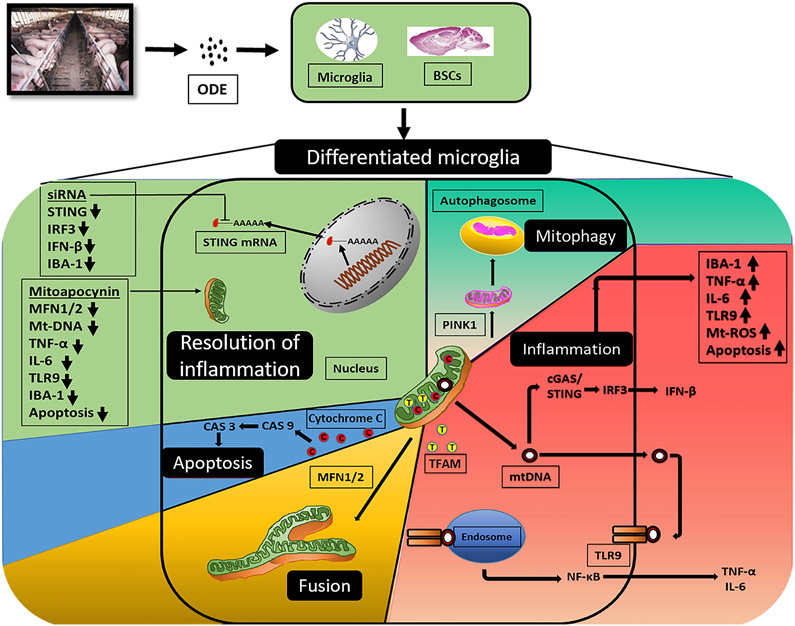
Organic dust (OD) exposure in animal production industries poses serious respiratory and other health risks. OD consisting of microbial products and particulate matter and OD exposure-induced respiratory inflammation are under investigation. However, the effect of OD exposure on brain remains elusive. We show that OD exposure of microglial cells induces an inflammatory phenotype with the release of mitochondrial DNA (mt-DNA). Therefore, we tested a hypothesis that OD exposure-induced secreted mt-DNA signaling drives the inflammation. A mouse microglial cell line was treated with medium or organic dust extract (ODE, 1% v/v) along with either phosphate-buffered saline (PBS) or mitoapocynin (MA, 10 µmol). Microglia treated with control or anti-STING siRNA were exposed to medium or ODE. Mouse organotypic brain slice cultures (BSCs) were exposed to medium or ODE with or without MA. Various samples were processed to quantify mitochondrial reactive oxygen species (mt-ROS), mt-DNA, cytochrome c, TFAM, mitochondrial stress markers and mt-DNA-induced signaling via cGAS-STING and TLR9. Data were analyzed and a p value of ≤ 0.05 was considered significant. MA treatment decreased the ODE-induced mt-DNA release into the cytosol. ODE increased MFN1/2 and PINK1 but not DRP1 and MA treatment decreased the MFN2 expression. MA treatment decreased the ODE exposure-induced mt-DNA signaling via cGAS-STING and TLR9. Anti-STING siRNA decreased the ODE-induced increase in IRF3, IFN-β and IBA-1 expression. In BSCs, MA treatment decreased the ODE-induced TNF-α, IL-6 and MFN1. Therefore, OD exposure-induced mt-DNA signaling was curtailed through cytoplasmic NOX-2 inhibition or STING suppression to reduce brain microglial inflammatory response.
Keywords: Inflammation; Microglia; Mitochondrial DNA; Organic dust; cGAS-STING.
Conflict of interest statement
Conflicts of interest/Competing interests
AGK has an equity interest in PK Biosciences Corporation located in Ames, IA.
The terms of this arrangement have been reviewed and approved by Iowa State University per its conflict of interest policies. All other authors have declared no potential conflicts of interest.
Figures
Similar articles
-
Organic dust exposure induces stress response and mitochondrial dysfunction in monocytic cells.Histochem Cell Biol. 2021 Jun;155(6):699-718. doi: 10.1007/s00418-021-01978-x. Epub 2021 Mar 23. Histochem Cell Biol. 2021. PMID: 33755775 Free PMC article.
-
HMGB1-RAGE Signaling Plays a Role in Organic Dust-Induced Microglial Activation and Neuroinflammation.Toxicol Sci. 2019 Jun 1;169(2):579-592. doi: 10.1093/toxsci/kfz071. Toxicol Sci. 2019. PMID: 30859215 Free PMC article.
-
Protective role of mitophagy on microglia-mediated neuroinflammatory injury through mtDNA-STING signaling in manganese-induced parkinsonism.J Neuroinflammation. 2025 Feb 28;22(1):55. doi: 10.1186/s12974-025-03396-5. J Neuroinflammation. 2025. PMID: 40022162 Free PMC article.
-
The cGAS-STING signaling in cardiovascular and metabolic diseases: Future novel target option for pharmacotherapy.Acta Pharm Sin B. 2022 Jan;12(1):50-75. doi: 10.1016/j.apsb.2021.05.011. Epub 2021 May 20. Acta Pharm Sin B. 2022. PMID: 35127372 Free PMC article. Review.
-
The role of cGAS-STING signalling in liver diseases.JHEP Rep. 2021 Jun 24;3(5):100324. doi: 10.1016/j.jhepr.2021.100324. eCollection 2021 Oct. JHEP Rep. 2021. PMID: 34381984 Free PMC article. Review.
Cited by
-
Mitochondrial DNA leakage: underlying mechanisms and therapeutic implications in neurological disorders.J Neuroinflammation. 2025 Feb 7;22(1):34. doi: 10.1186/s12974-025-03363-0. J Neuroinflammation. 2025. PMID: 39920753 Free PMC article. Review.
-
Mitochondrial dysfunction in chronic neuroinflammatory diseases (Review).Int J Mol Med. 2024 May;53(5):47. doi: 10.3892/ijmm.2024.5371. Epub 2024 Apr 5. Int J Mol Med. 2024. PMID: 38577947 Free PMC article. Review.
-
The NADPH Oxidase Inhibitor, Mitoapocynin, Mitigates DFP-Induced Reactive Astrogliosis in a Rat Model of Organophosphate Neurotoxicity.Antioxidants (Basel). 2023 Nov 30;12(12):2061. doi: 10.3390/antiox12122061. Antioxidants (Basel). 2023. PMID: 38136181 Free PMC article.
-
Transcriptomic and ultrastructural evidence indicate that anti-HMGB1 antibodies rescue organic dust-induced mitochondrial dysfunction.Cell Tissue Res. 2022 May;388(2):373-398. doi: 10.1007/s00441-022-03602-3. Epub 2022 Mar 4. Cell Tissue Res. 2022. PMID: 35244775 Free PMC article.
-
Mitoapocynin Attenuates Organic Dust Exposure-Induced Neuroinflammation and Sensory-Motor Deficits in a Mouse Model.Front Cell Neurosci. 2022 Apr 13;16:817046. doi: 10.3389/fncel.2022.817046. eCollection 2022. Front Cell Neurosci. 2022. PMID: 35496912 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
