

Therapeutic Targeting of the Complement System: From Rare Diseases to Pandemics
- PMID: 33687995
- PMCID: PMC7956994
- DOI: 10.1124/pharmrev.120.000072
Therapeutic Targeting of the Complement System: From Rare Diseases to Pandemics
Abstract
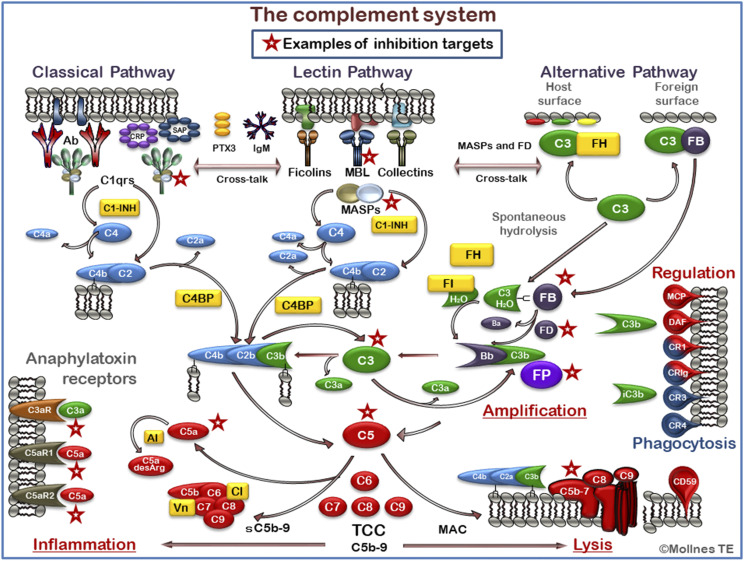
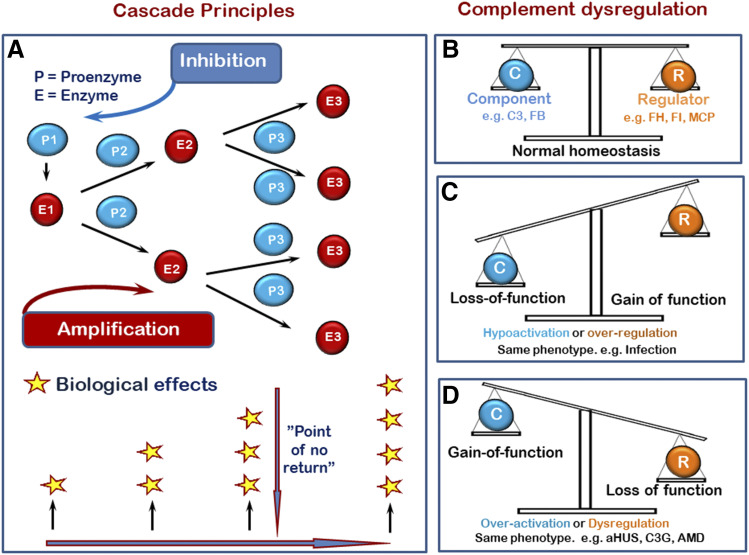
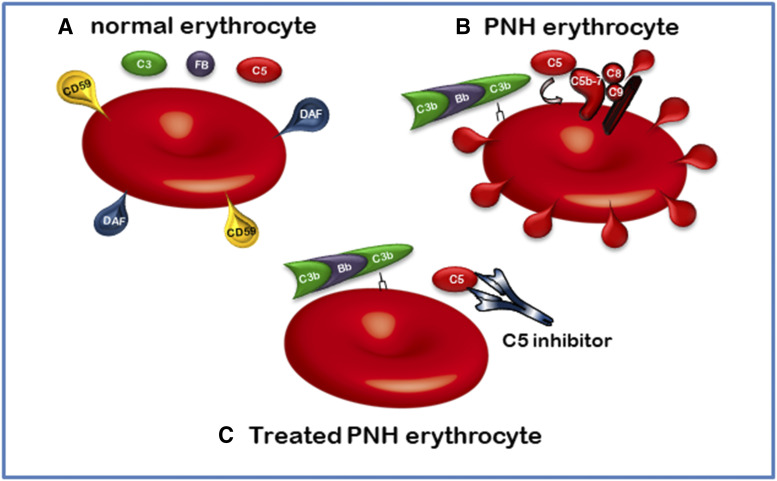
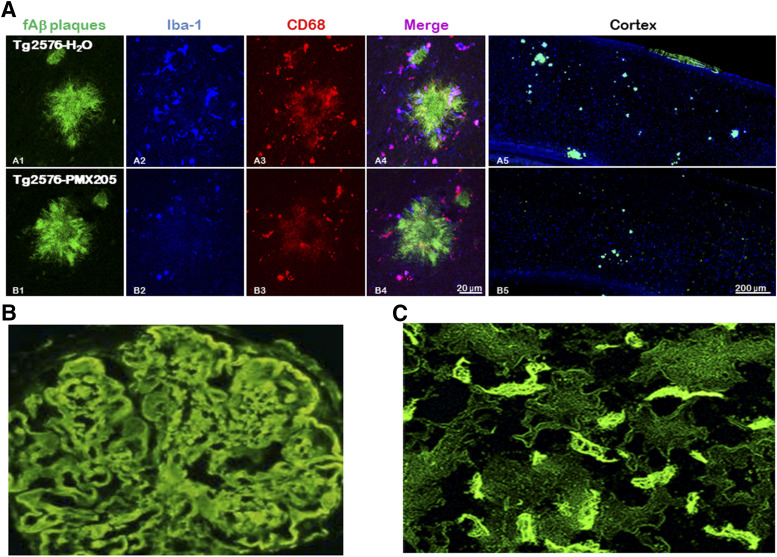
The complement system was discovered at the end of the 19th century as a heat-labile plasma component that "complemented" the antibodies in killing microbes, hence the name "complement." Complement is also part of the innate immune system, protecting the host by recognition of pathogen-associated molecular patterns. However, complement is multifunctional far beyond infectious defense. It contributes to organ development, such as sculpting neuron synapses, promoting tissue regeneration and repair, and rapidly engaging and synergizing with a number of processes, including hemostasis leading to thromboinflammation. Complement is a double-edged sword. Although it usually protects the host, it may cause tissue damage when dysregulated or overactivated, such as in the systemic inflammatory reaction seen in trauma and sepsis and severe coronavirus disease 2019 (COVID-19). Damage-associated molecular patterns generated during ischemia-reperfusion injuries (myocardial infarction, stroke, and transplant dysfunction) and in chronic neurologic and rheumatic disease activate complement, thereby increasing damaging inflammation. Despite the long list of diseases with potential for ameliorating complement modulation, only a few rare diseases are approved for clinical treatment targeting complement. Those currently being efficiently treated include paroxysmal nocturnal hemoglobinuria, atypical hemolytic-uremic syndrome, myasthenia gravis, and neuromyelitis optica spectrum disorders. Rare diseases, unfortunately, preclude robust clinical trials. The increasing evidence for complement as a pathogenetic driver in many more common diseases suggests an opportunity for future complement therapy, which, however, requires robust clinical trials; one ongoing example is COVID-19 disease. The current review aims to discuss complement in disease pathogenesis and discuss future pharmacological strategies to treat these diseases with complement-targeted therapies. SIGNIFICANCE STATEMENT: The complement system is the host's defense friend by protecting it from invading pathogens, promoting tissue repair, and maintaining homeostasis. Complement is a double-edged sword, since when dysregulated or overactivated it becomes the host's enemy, leading to tissue damage, organ failure, and, in worst case, death. A number of acute and chronic diseases are candidates for pharmacological treatment to avoid complement-dependent damage, ranging from the well established treatment for rare diseases to possible future treatment of large patient groups like the pandemic coronavirus disease 2019.
Copyright © 2021 by The Author(s).
Conflict of interest statement
Conflict of Interest: T.E.M. is a member of the Scientific Advisory Board of Ra Pharma/UCB. A.J.T. is a consultant for Montis.
Figures

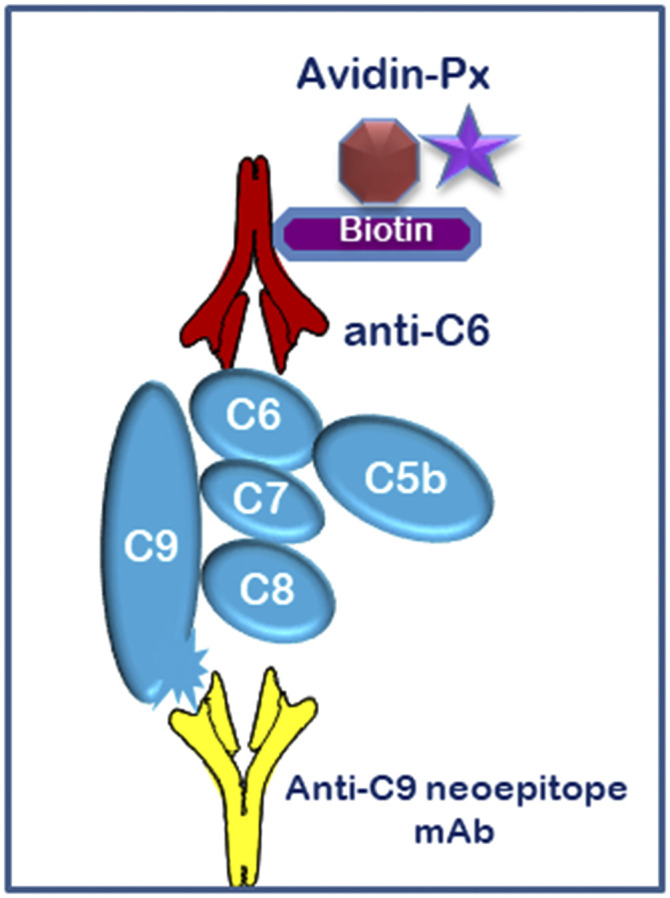
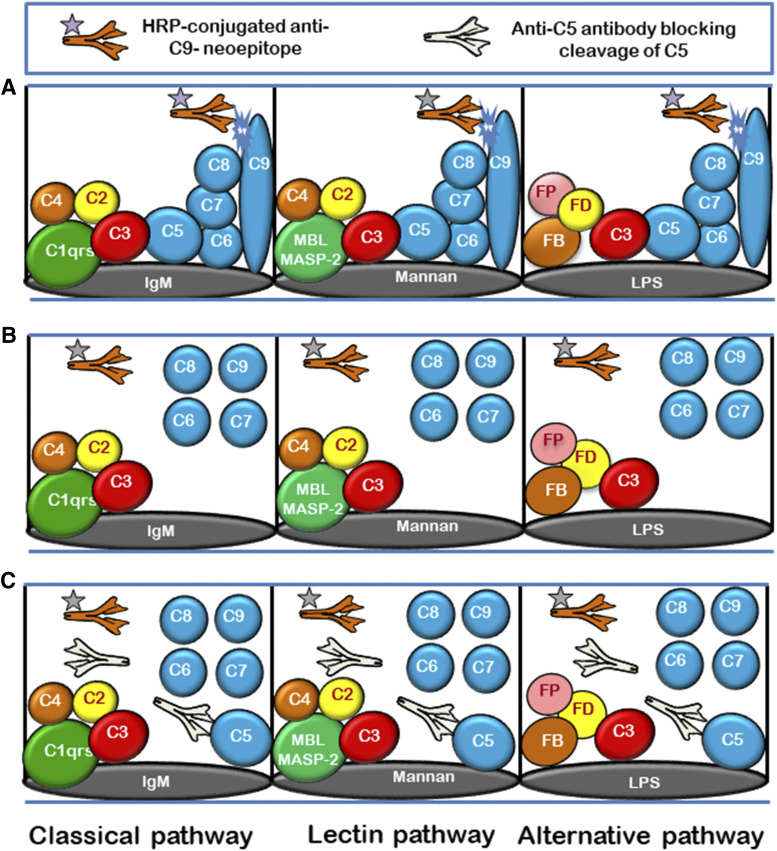
References
-
- Ahmad SB, Bomback AS (2020) C3 glomerulopathy: pathogenesis and treatment. Adv Chronic Kidney Dis 27:104–110. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
