

Polymicrobial communities in periodontal disease: Their quasi-organismal nature and dialogue with the host
- PMID: 33690950
- PMCID: PMC8957750
- DOI: 10.1111/prd.12371
Polymicrobial communities in periodontal disease: Their quasi-organismal nature and dialogue with the host
Abstract
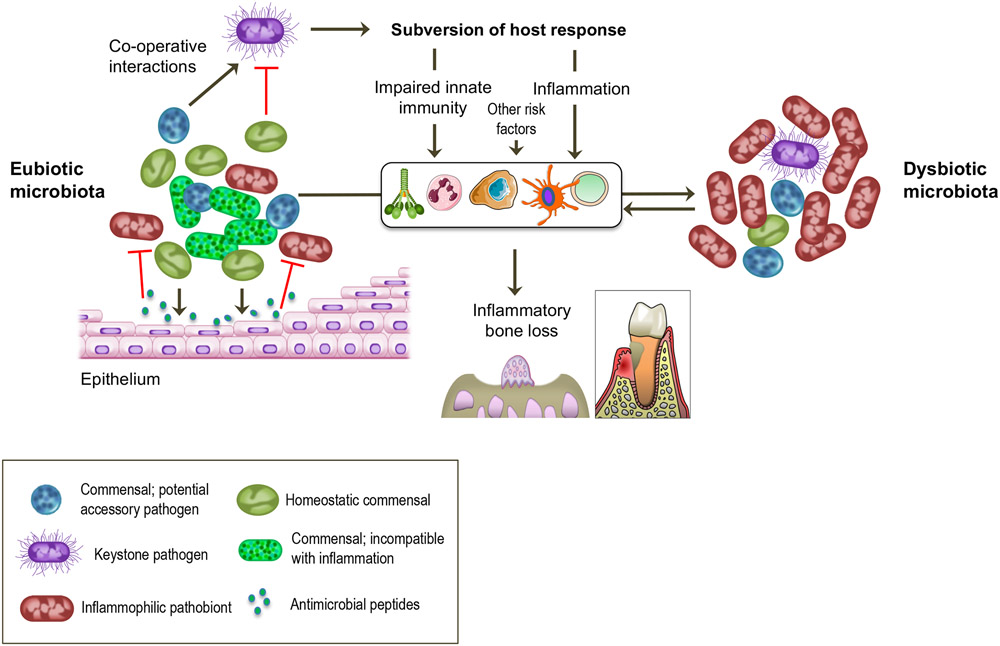
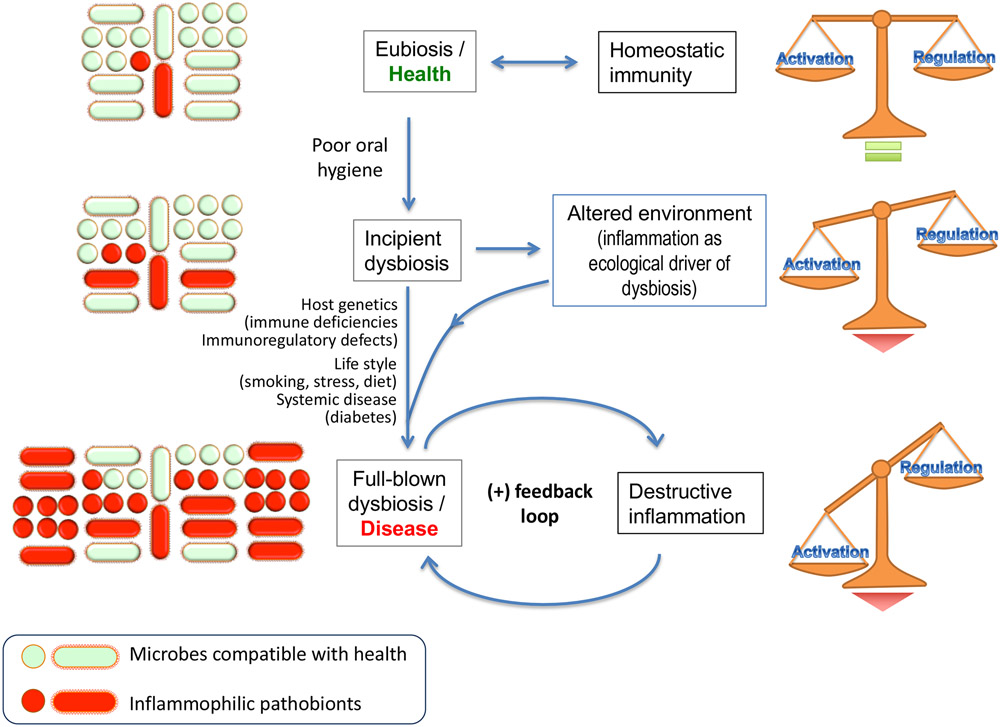
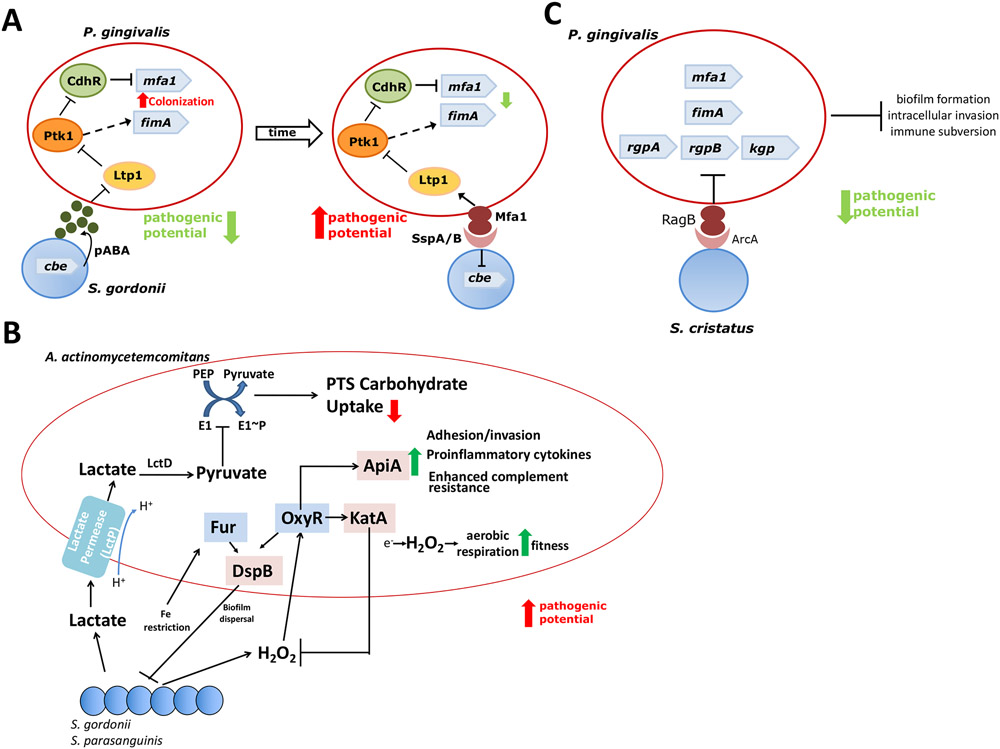
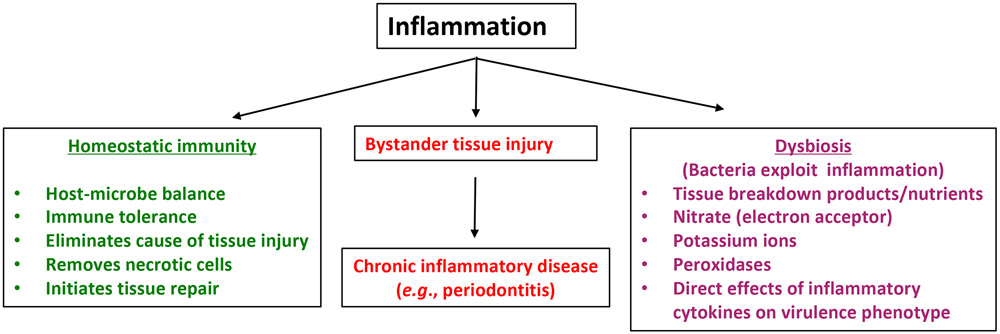
In health, indigenous polymicrobial communities at mucosal surfaces maintain an ecological balance via both inter-microbial and host-microbial interactions that promote their own and the host's fitness, while preventing invasion by exogenous pathogens. However, genetic and acquired destabilizing factors (including immune deficiencies, immunoregulatory defects, smoking, diet, obesity, diabetes and other systemic diseases, and aging) may disrupt this homeostatic balance, leading to selective outgrowth of species with the potential for destructive inflammation. This process, known as dysbiosis, underlies the development of periodontitis in susceptible hosts. The pathogenic process is not linear but involves a positive-feedback loop between dysbiosis and the host inflammatory response. The dysbiotic community is essentially a quasi-organismal entity, where constituent organisms communicate via sophisticated physical and chemical signals and display functional specialization (eg, accessory pathogens, keystone pathogens, pathobionts), which enables polymicrobial synergy and dictates the community's pathogenic potential or nososymbiocity. In this review, we discuss early and recent studies in support of the polymicrobial synergy and dysbiosis model of periodontal disease pathogenesis. According to this concept, disease is not caused by individual "causative pathogens" but rather by reciprocally reinforced interactions between physically and metabolically integrated polymicrobial communities and a dysregulated host inflammatory response.
Keywords: dysbiosis; inflammation; keystone pathogen; pathobiont; periodontitis; polymicrobial community.
© 2021 John Wiley & Sons A/S. Published by John Wiley & Sons Ltd.
Figures
References
-
- Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol. 2010;8(7):481–490. - PubMed
Publication types
MeSH terms
Grants and funding
- DE017921/NH/NIH HHS/United States
- GM125504/NH/NIH HHS/United States
- R37 DE026152/DE/NIDCR NIH HHS/United States
- R01 DE023193/DE/NIDCR NIH HHS/United States
- DE028561/NH/NIH HHS/United States
- R37 DE011111/DE/NIDCR NIH HHS/United States
- DE024153/NH/NIH HHS/United States
- DE012505/NH/NIH HHS/United States
- DE024716/NH/NIH HHS/United States
- DE029436/NH/NIH HHS/United States
- R01 DE024716/DE/NIDCR NIH HHS/United States
- R01 DE029436/DE/NIDCR NIH HHS/United States
- P20 GM125504/GM/NIGMS NIH HHS/United States
- R01 DE028561/DE/NIDCR NIH HHS/United States
- R01 DE012505/DE/NIDCR NIH HHS/United States
- DE023193/NH/NIH HHS/United States
- DE01111/NH/NIH HHS/United States
- R01 DE017921/DE/NIDCR NIH HHS/United States
- DE026152/NH/NIH HHS/United States
- R01 DE024153/DE/NIDCR NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
