

Most non-canonical proteins uniquely populate the proteome or immunopeptidome
- PMID: 33691108
- PMCID: PMC8040094
- DOI: 10.1016/j.celrep.2021.108815
Most non-canonical proteins uniquely populate the proteome or immunopeptidome
Abstract
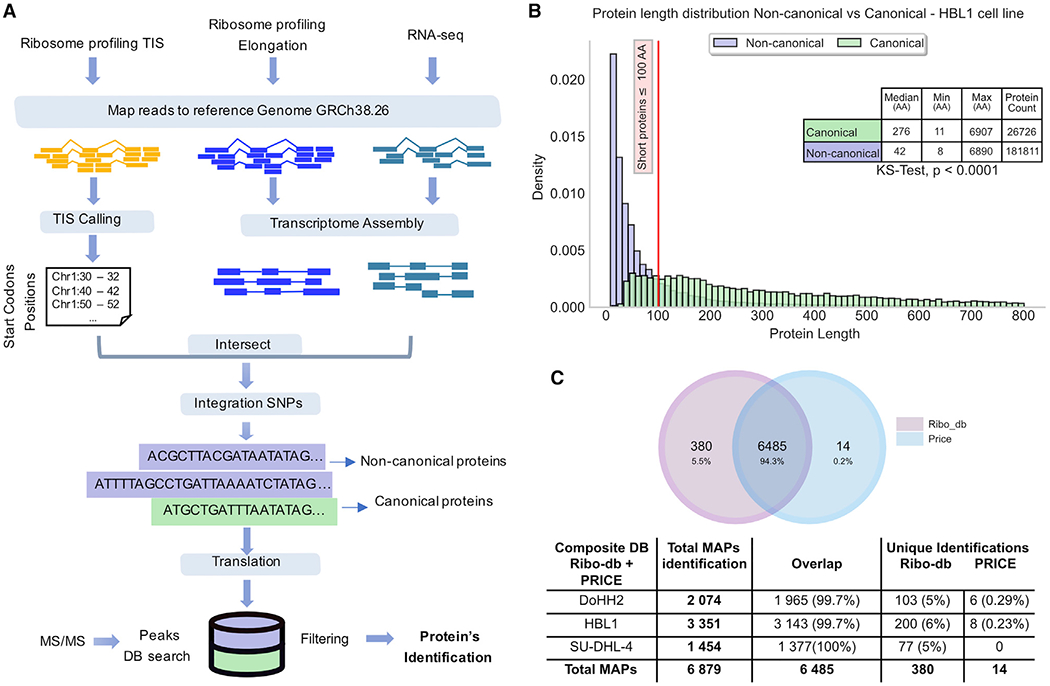
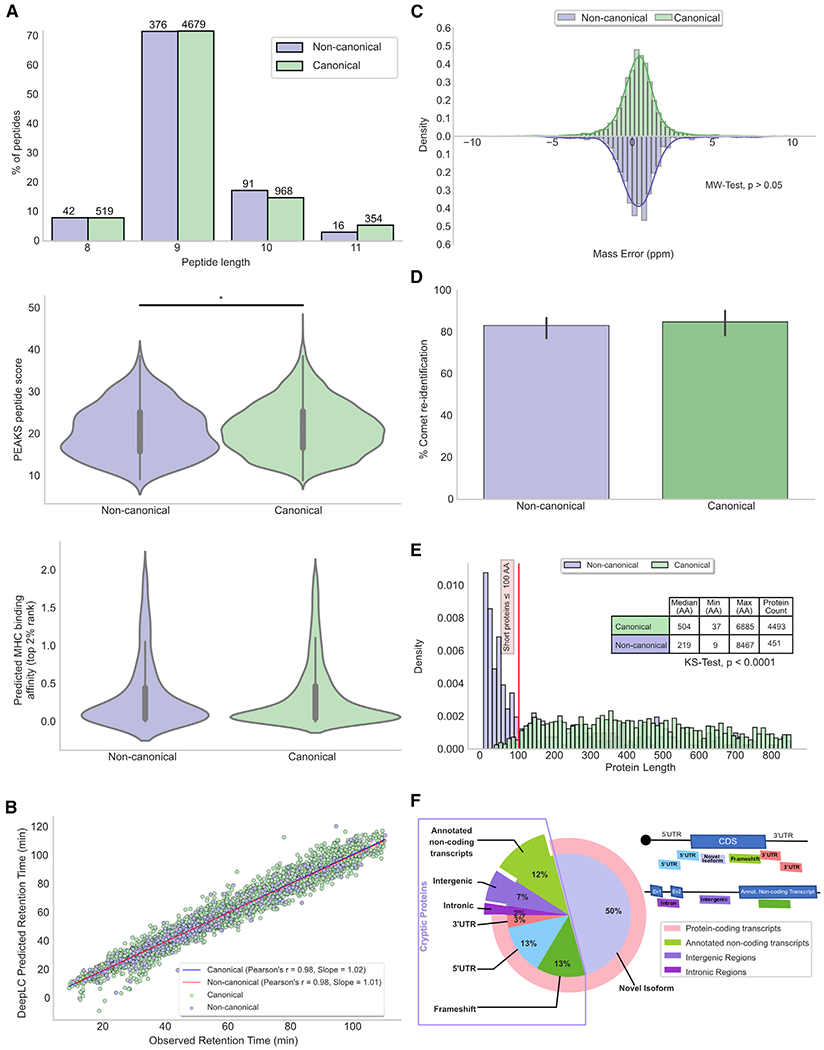
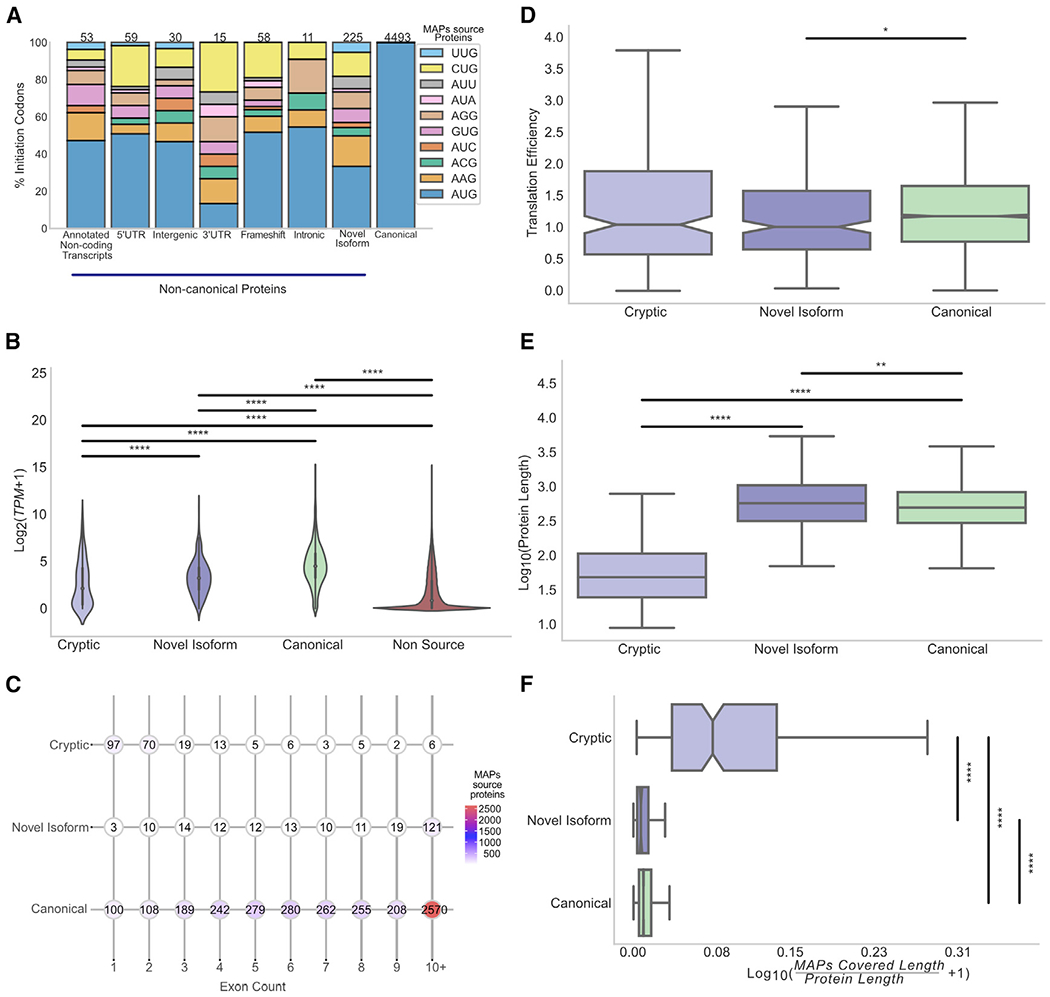
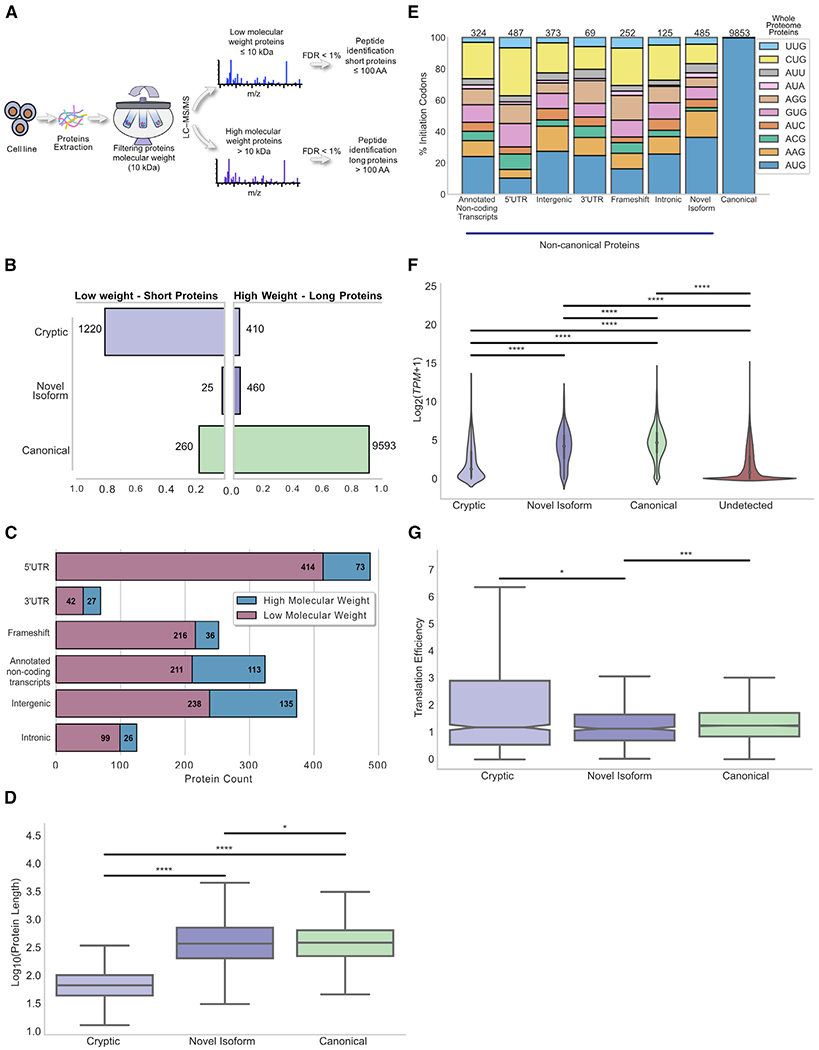
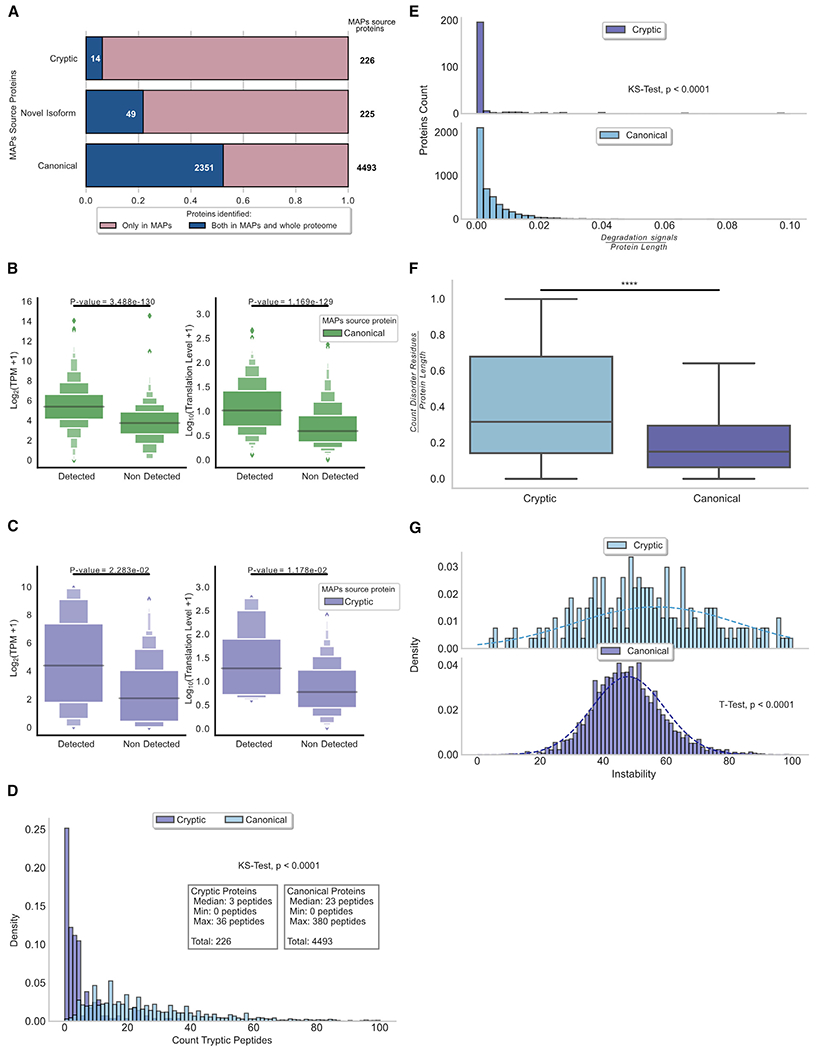
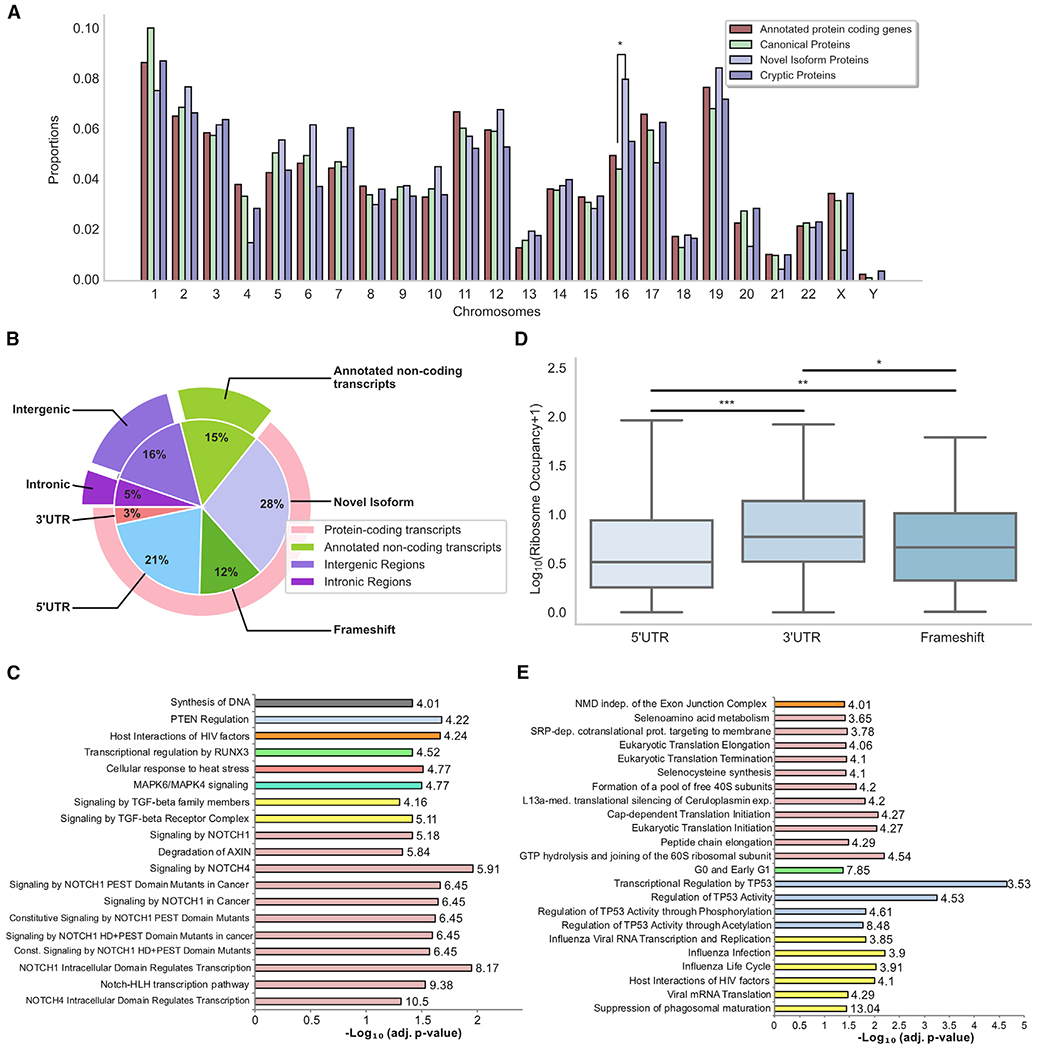
Combining RNA sequencing, ribosome profiling, and mass spectrometry, we elucidate the contribution of non-canonical translation to the proteome and major histocompatibility complex (MHC) class I immunopeptidome. Remarkably, of 14,498 proteins identified in three human B cell lymphomas, 2,503 are non-canonical proteins. Of these, 28% are novel isoforms and 72% are cryptic proteins encoded by ostensibly non-coding regions (60%) or frameshifted canonical genes (12%). Cryptic proteins are translated as efficiently as canonical proteins, have more predicted disordered residues and lower stability, and critically generate MHC-I peptides 5-fold more efficiently per translation event. Translating 5' "untranslated" regions hinders downstream translation of genes involved in transcription, translation, and antiviral responses. Novel protein isoforms show strong enrichment for signaling pathways deregulated in cancer. Only a small fraction of cryptic proteins detected in the proteome contribute to the MHC-I immunopeptidome, demonstrating the high preferential access of cryptic defective ribosomal products to the class I pathway.
Keywords: computational biology; defective ribosomal products; major histocompatibility complex; mass spectrometry; non-canonical translation; peptides; protein isoforms; proteomic methods; ribosome profiling.
Copyright © 2021 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
