

Machine Learning Identifies Complicated Sepsis Course and Subsequent Mortality Based on 20 Genes in Peripheral Blood Immune Cells at 24 H Post-ICU Admission
- PMID: 33692779
- PMCID: PMC7937924
- DOI: 10.3389/fimmu.2021.592303
Machine Learning Identifies Complicated Sepsis Course and Subsequent Mortality Based on 20 Genes in Peripheral Blood Immune Cells at 24 H Post-ICU Admission
Abstract
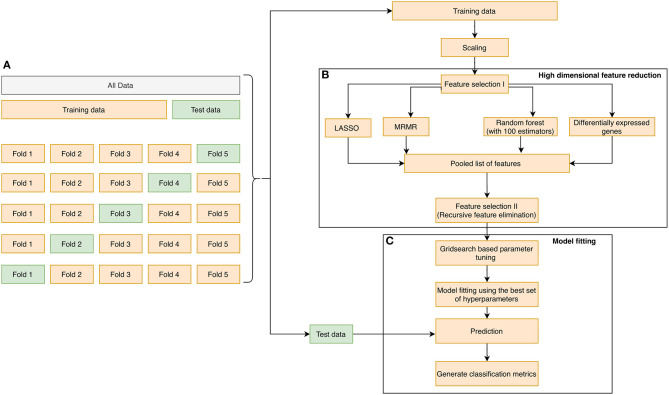
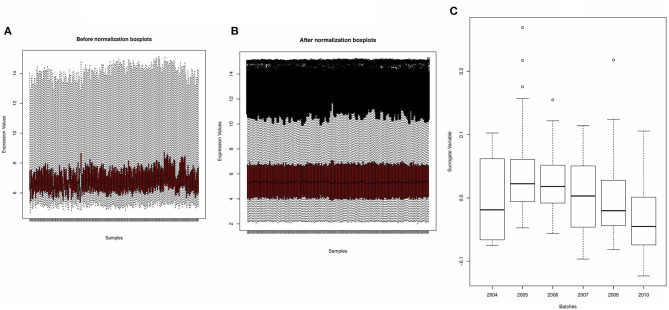
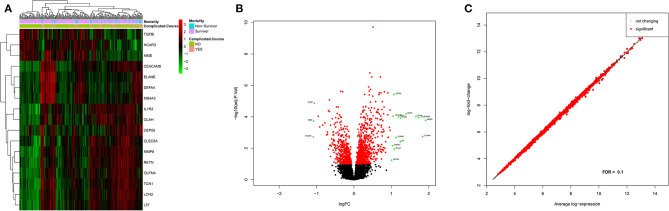
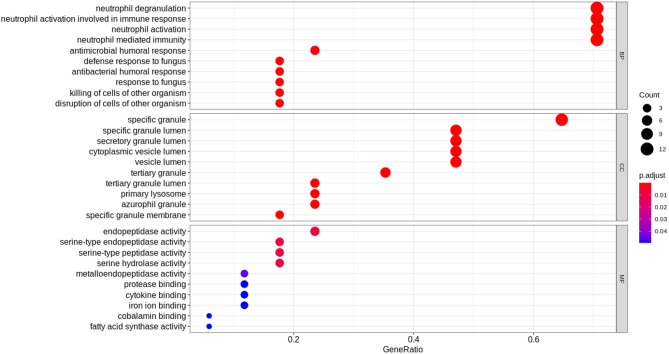
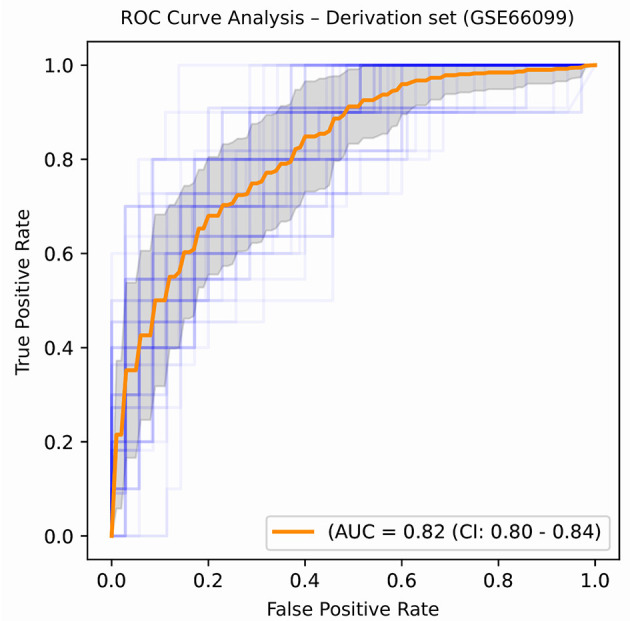
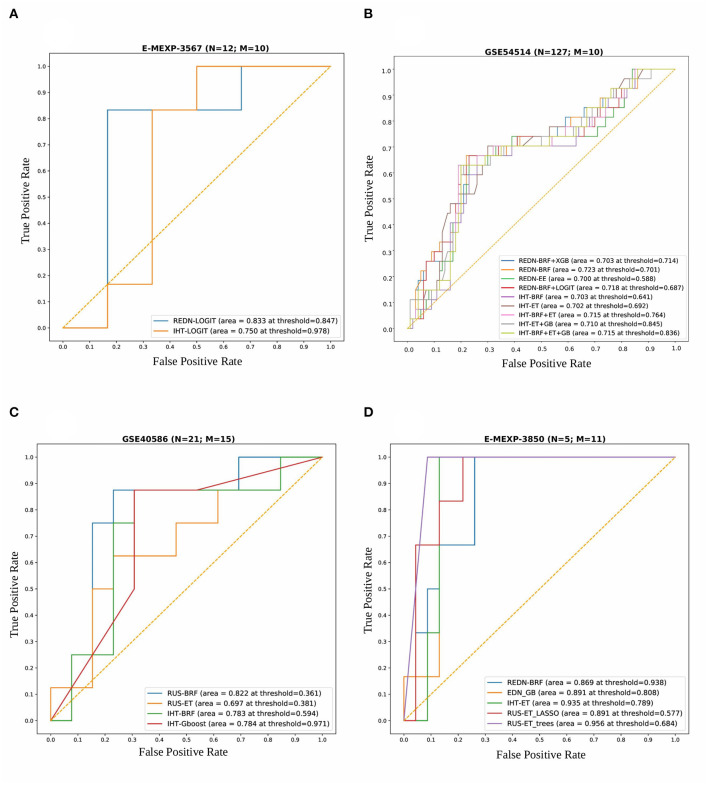
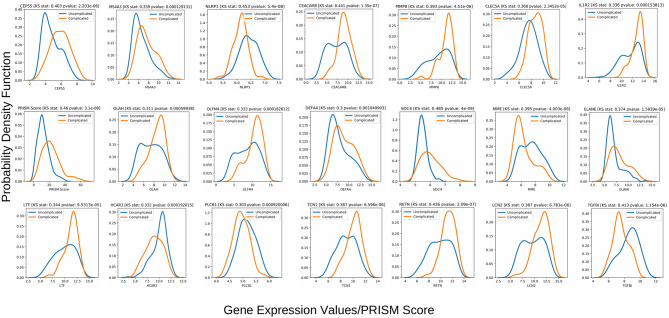
A complicated clinical course for critically ill patients admitted to the intensive care unit (ICU) usually includes multiorgan dysfunction and subsequent death. Owing to the heterogeneity, complexity, and unpredictability of the disease progression, ICU patient care is challenging. Identifying the predictors of complicated courses and subsequent mortality at the early stages of the disease and recognizing the trajectory of the disease from the vast array of longitudinal quantitative clinical data is difficult. Therefore, we attempted to perform a meta-analysis of previously published gene expression datasets to identify novel early biomarkers and train the artificial intelligence systems to recognize the disease trajectories and subsequent clinical outcomes. Using the gene expression profile of peripheral blood cells obtained within 24 h of pediatric ICU (PICU) admission and numerous clinical data from 228 septic patients from pediatric ICU, we identified 20 differentially expressed genes predictive of complicated course outcomes and developed a new machine learning model. After 5-fold cross-validation with 10 iterations, the overall mean area under the curve reached 0.82. Using a subset of the same set of genes, we further achieved an overall area under the curve of 0.72, 0.96, 0.83, and 0.82, respectively, on four independent external validation sets. This model was highly effective in identifying the clinical trajectories of the patients and mortality. Artificial intelligence systems identified eight out of twenty novel genetic markers (SDC4, CLEC5A, TCN1, MS4A3, HCAR3, OLAH, PLCB1, and NLRP1) that help predict sepsis severity or mortality. While these genes have been previously associated with sepsis mortality, in this work, we show that these genes are also implicated in complex disease courses, even among survivors. The discovery of eight novel genetic biomarkers related to the overactive innate immune system, including neutrophil function, and a new predictive machine learning method provides options to effectively recognize sepsis trajectories, modify real-time treatment options, improve prognosis, and patient survival.
Keywords: biomarkers; complicated course; critical care; machine learning; sepsis; transcriptomics.
Copyright © 2021 Banerjee, Mohammed, Wong, Palaniyar and Kamaleswaran.
Conflict of interest statement
HW and Cincinnati Children's Hospital Medical Center hold United States patents for the PERSEVERE biomarkers and the endotyping strategy described in this manuscript. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical