

IPEX Syndrome: Improved Knowledge of Immune Pathogenesis Empowers Diagnosis
- PMID: 33692972
- PMCID: PMC7937806
- DOI: 10.3389/fped.2021.612760
IPEX Syndrome: Improved Knowledge of Immune Pathogenesis Empowers Diagnosis
Abstract
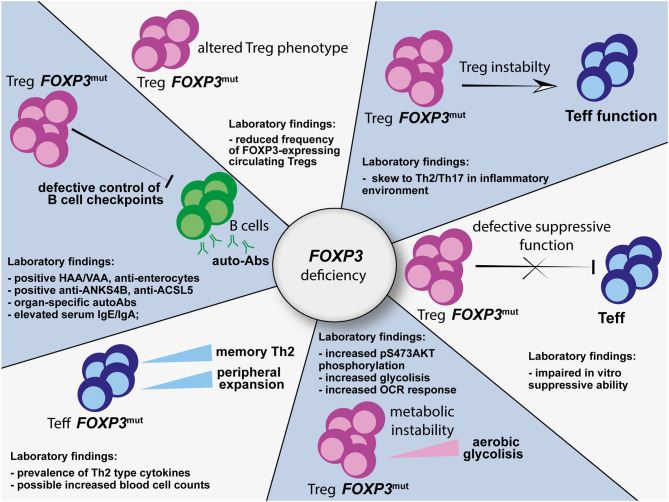
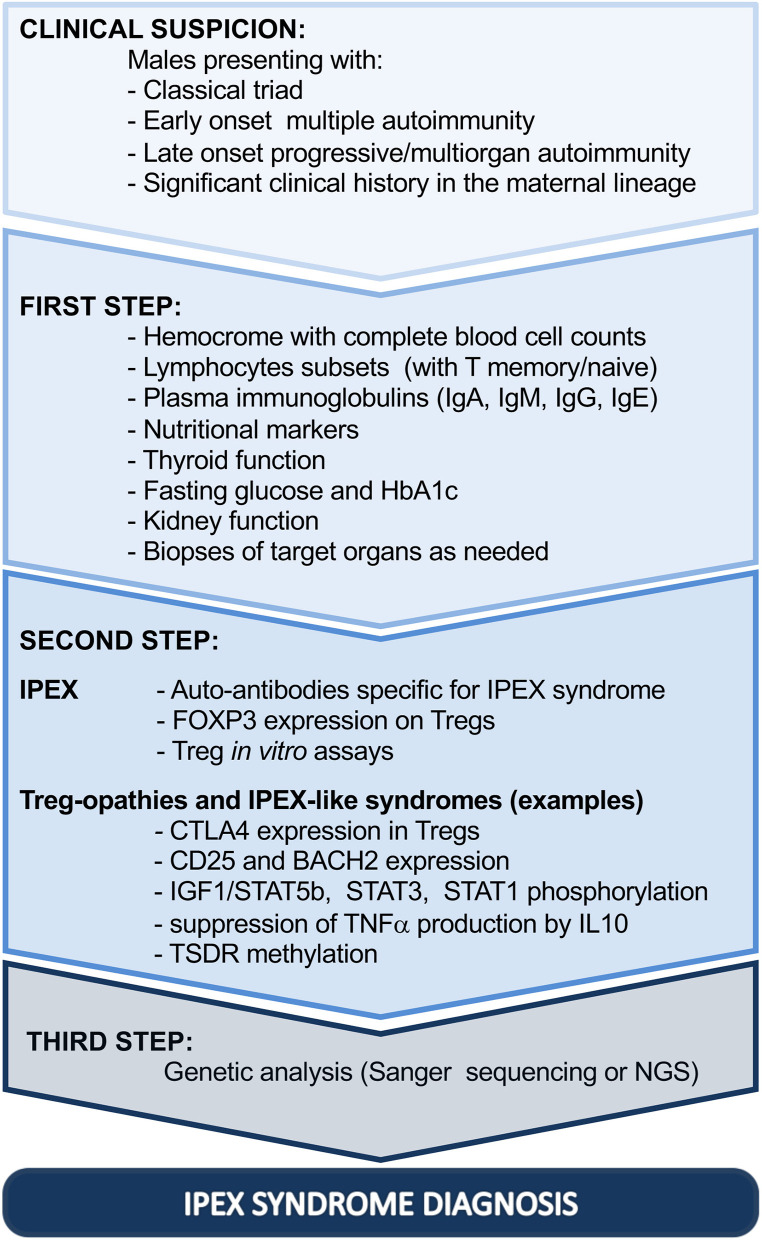
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome is a rare monogenic autoimmune disease with variable clinical manifestations, ranging from early-onset severe autoimmunity, including enteropathy, eczema, and type 1 diabetes, to late-onset or atypical symptoms. Despite the clinical heterogeneity, the unifying feature of IPEX is mutation of the FOXP3 gene, which encodes a transcription factor essential for maintenance of thymus-derived regulatory T cells (Tregs). In IPEX patients, Tregs can be present, although unstable and impaired in function, unable to inhibit proliferation and cytokine production of effector T (Teff) cells. Mutated FOXP3 can also disrupt other compartments: FOXP3-deficient Teff cells proliferate more than the wild-type counterpart, display altered T-cell-receptor signaling response, a reduced T-naïve compartment and a skew toward a Th2 profile. Due to FOXP3 mutations, the frequency of autoreactive B cells is increased and the IgA and IgE production is altered, together with early emergence of tissue-specific autoantibodies. Recently, the awareness of the wide clinical spectrum of IPEX improved the diagnostic tools. In cases presenting with enteropathy, histological evaluation is helpful, although there are no pathognomonic signs of disease. On the other hand, the study of FOXP3 expression and in vitro Treg function, as well as the detection of specific circulating autoantibodies, is recommended to narrow the differential diagnosis. Nowadays, Sanger sequencing should be limited to cases presenting with the classical triad of symptoms; otherwise, next-generation sequencing is recommended, given the cost-effectiveness and the advantage of excluding IPEX-like syndromes. The latter approach could be time spearing in children with severe phenotypes and candidate to advanced therapies.
Keywords: FOXP3; IPEX syndrome; autoimmunity; diagnosis; immune tolerance; next generation sequencing; regulatory T cells.
Copyright © 2021 Barzaghi and Passerini.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Powell BR, Buist NR, Stenzel P. An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. Journal Pediatr. (1982) 100:731–7. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous