

The GGC repeat expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy type 3
- PMID: 33693509
- PMCID: PMC8320266
- DOI: 10.1093/brain/awab077
The GGC repeat expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy type 3
Erratum in
-
Erratum to: The GGC repeat expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy type 3.Brain. 2021 Oct 22;144(9):e81. doi: 10.1093/brain/awab266. Brain. 2021. PMID: 34435201 Free PMC article. No abstract available.
Abstract
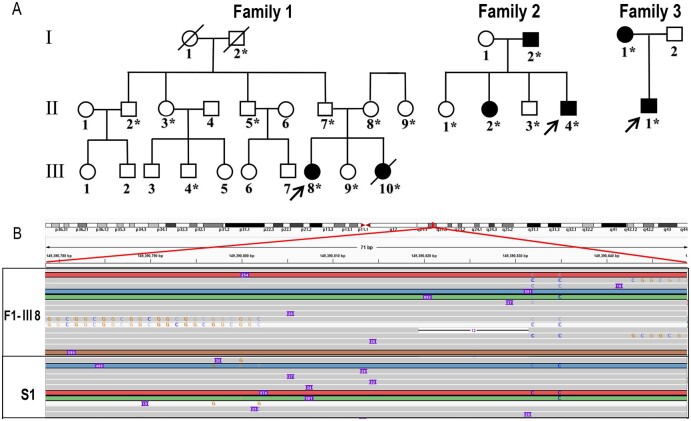
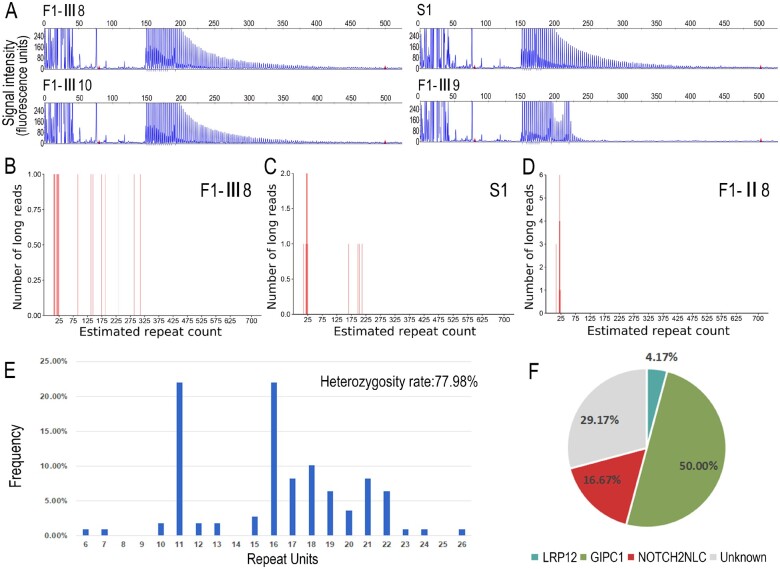
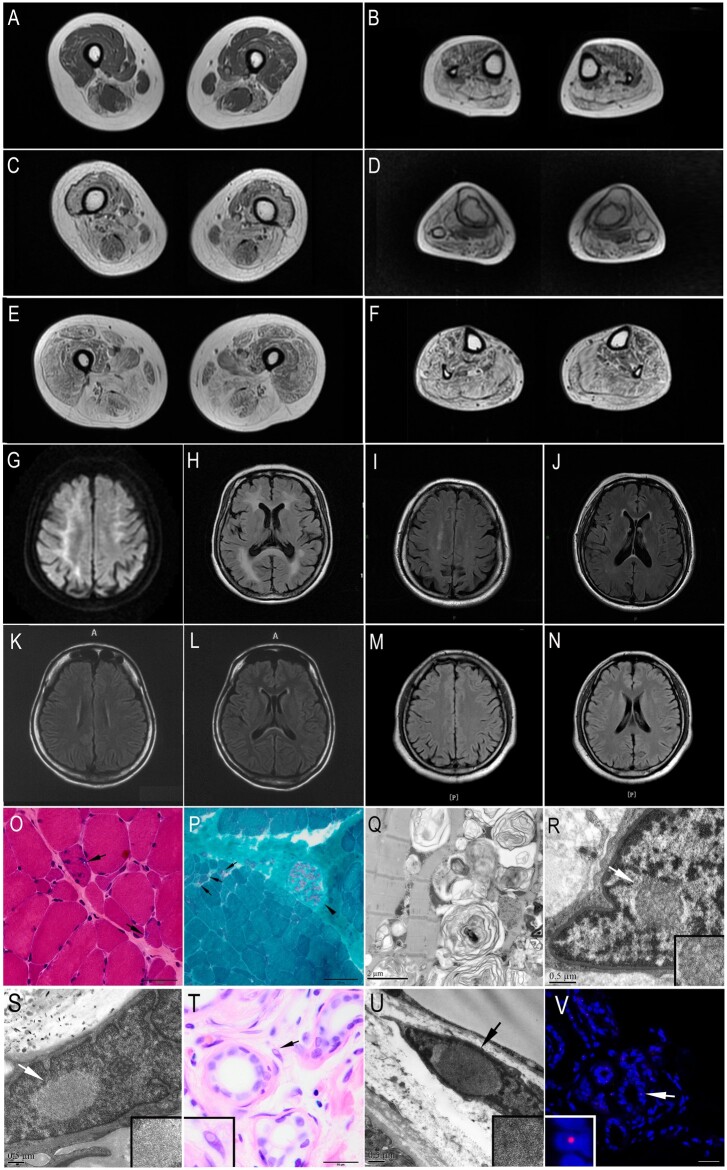
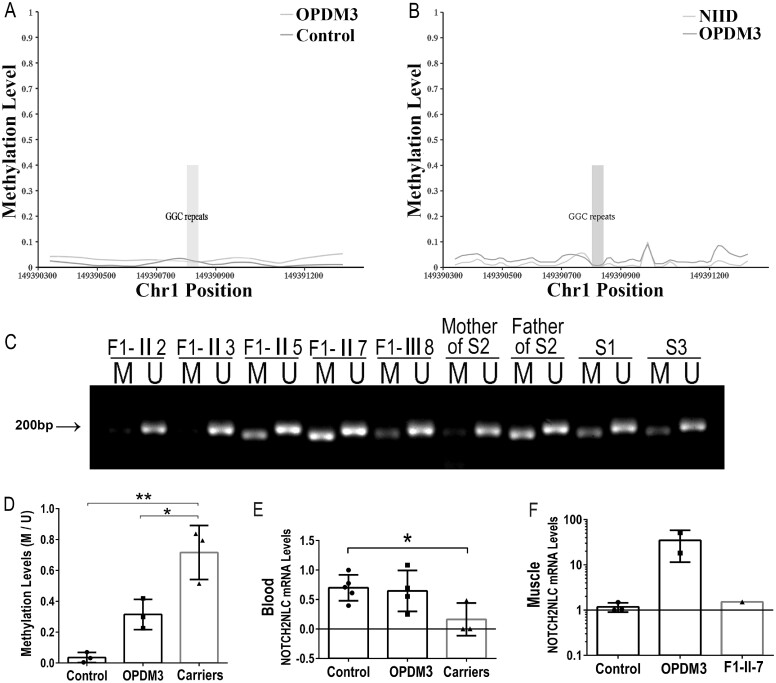
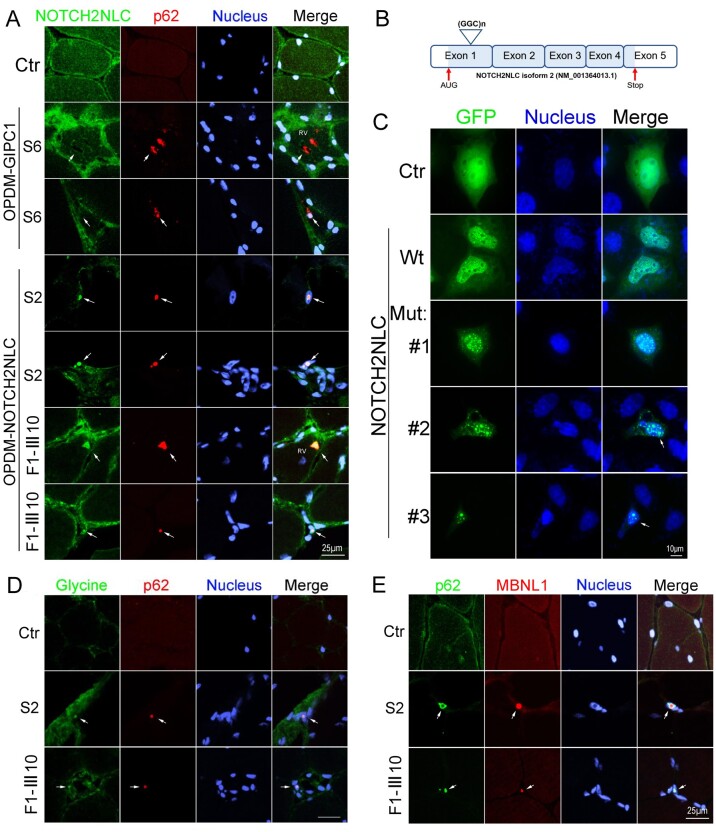
Oculopharyngodistal myopathy (OPDM) is an adult-onset neuromuscular disease characterized by progressive ocular, facial, pharyngeal and distal limb muscle involvement. Trinucleotide repeat expansions in LRP12 or GIPC1 were recently reported to be associated with OPDM. However, a significant portion of OPDM patients have unknown genetic causes. In this study, long-read whole-genome sequencing and repeat-primed PCR were performed and we identified GGC repeat expansions in the NOTCH2NLC gene in 16.7% (4/24) of a cohort of Chinese OPDM patients, designated as OPDM type 3 (OPDM3). Methylation analysis indicated that methylation levels of the NOTCH2NLC gene were unaltered in OPDM3 patients, but increased significantly in asymptomatic carriers. Quantitative real-time PCR analysis indicated that NOTCH2NLC mRNA levels were increased in muscle but not in blood of OPDM3 patients. Immunofluorescence on OPDM muscle samples and expressing mutant NOTCH2NLC with (GGC)69 repeat expansions in HEK293 cells indicated that mutant NOTCH2NLC-polyglycine protein might be a major component of intranuclear inclusions, and contribute to toxicity in cultured cells. In addition, two RNA-binding proteins, hnRNP A/B and MBNL1, were both co-localized with p62 in intranuclear inclusions in OPDM muscle samples. These results indicated that a toxic protein gain-of-function mechanism and RNA gain-of-function mechanism may both play a vital role in the pathogenic processes of OPDM3. This study extended the spectrum of NOTCH2NLC repeat expansion-related diseases to a predominant myopathy phenotype presenting as OPDM, and provided evidence for possible pathogenesis of these diseases.
Keywords: GGC repeat expansion; NOTCH2NLC; RNA gain-of-function mechanism; oculopharyngodistal myopathy; toxic protein gain-of-function mechanism.
© The Author(s) (2021). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
References
-
- Durmus H, Laval SH, Deymeer F, et al. Oculopharyngodistal myopathy is a distinct entity: Clinical and genetic features of 47 patients. Neurology. 2011;76:227–235. - PubMed
-
- Minami N, Ikezoe K, Kuroda H, Nakabayashi H, Satoyoshi E, Nonaka IJND.. Oculopharyngodistal myopathy is genetically heterogeneous and most cases are distinct from oculopharyngeal muscular dystrophy. Neuromuscul Disord. 2001;11:699–702. - PubMed
-
- Lu H, Luan X, Yuan Y, Dong M, Sun W, Yan C.. The clinical and myopathological features of oculopharyngodistal myopathy in a Chinese family. Neuropathology. 2008;28:599–603. - PubMed
-
- Satoyoshi E, Kinoshita M.. Oculopharyngodistal myopathy. Arch Neurol. 1977;34:89–92. - PubMed
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
