

Protein interaction interface region prediction by geometric deep learning
- PMID: 33693581
- PMCID: PMC8428585
- DOI: 10.1093/bioinformatics/btab154
Protein interaction interface region prediction by geometric deep learning
Abstract
Motivation: Protein-protein interactions drive wide-ranging molecular processes, and characterizing at the atomic level how proteins interact (beyond just the fact that they interact) can provide key insights into understanding and controlling this machinery. Unfortunately, experimental determination of three-dimensional protein complex structures remains difficult and does not scale to the increasingly large sets of proteins whose interactions are of interest. Computational methods are thus required to meet the demands of large-scale, high-throughput prediction of how proteins interact, but unfortunately, both physical modeling and machine learning methods suffer from poor precision and/or recall.
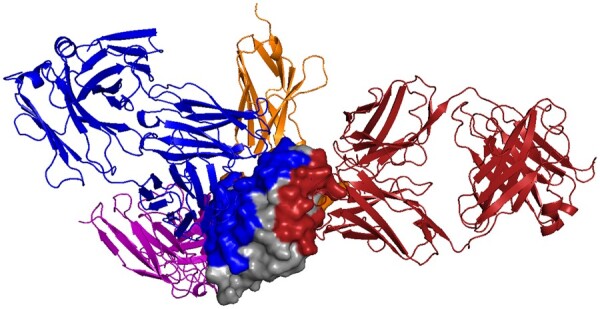
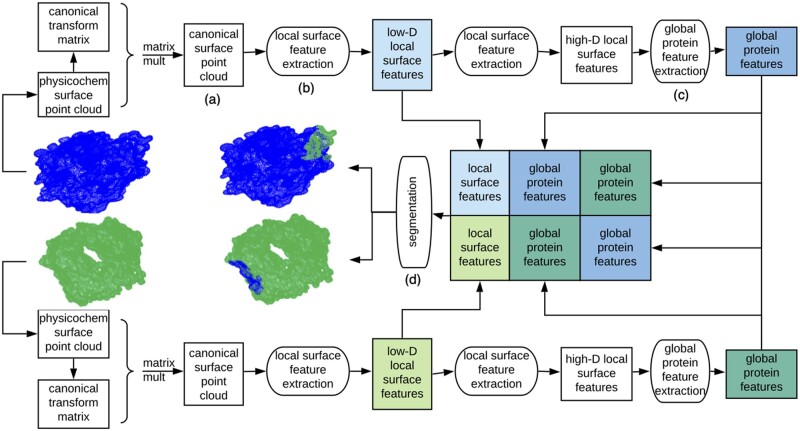
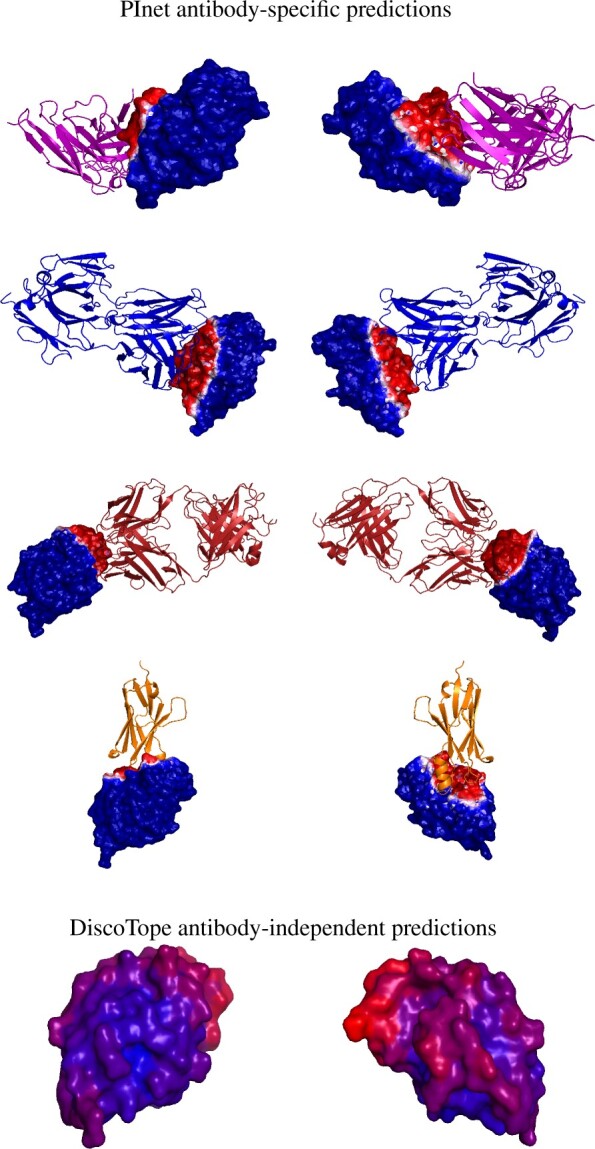
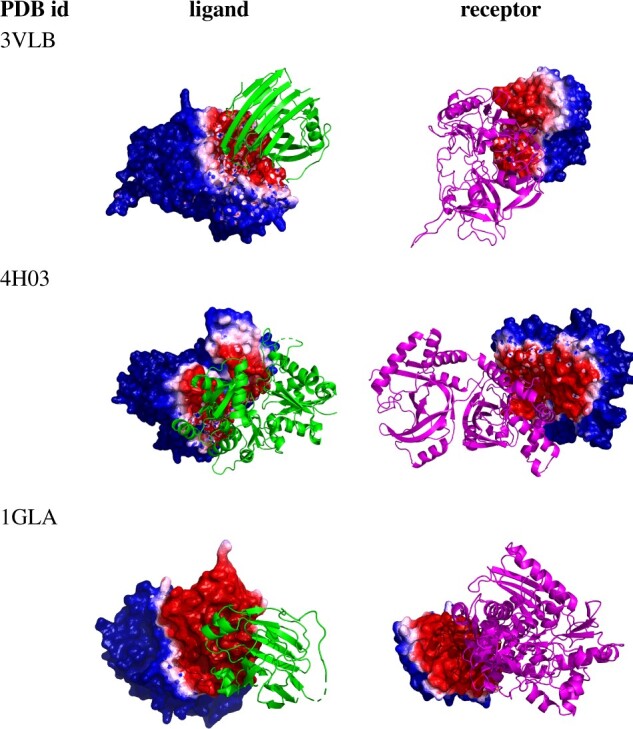
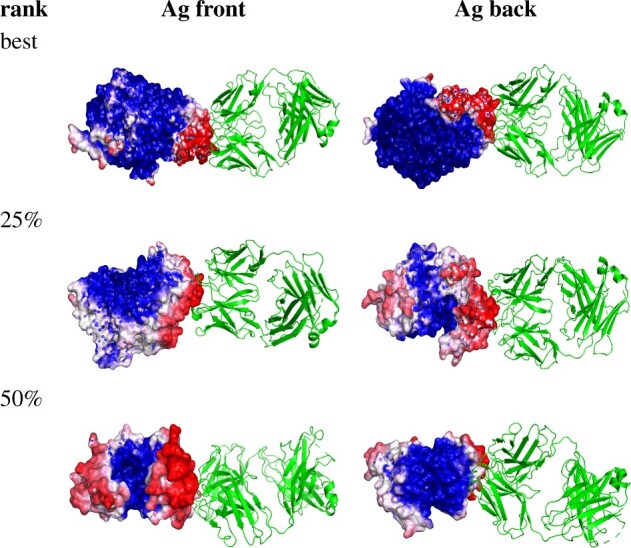
Results: In order to improve performance in predicting protein interaction interfaces, we leverage the best properties of both data- and physics-driven methods to develop a unified Geometric Deep Neural Network, 'PInet' (Protein Interface Network). PInet consumes pairs of point clouds encoding the structures of two partner proteins, in order to predict their structural regions mediating interaction. To make such predictions, PInet learns and utilizes models capturing both geometrical and physicochemical molecular surface complementarity. In application to a set of benchmarks, PInet simultaneously predicts the interface regions on both interacting proteins, achieving performance equivalent to or even much better than the state-of-the-art predictor for each dataset. Furthermore, since PInet is based on joint segmentation of a representation of a protein surfaces, its predictions are meaningful in terms of the underlying physical complementarity driving molecular recognition.
Availability and implementation: PInet scripts and models are available at https://github.com/FTD007/PInet.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2021. Published by Oxford University Press. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
References
-
- Bahdanau D. et al. (2014) Neural machine translation by jointly learning to align and translate. arXiv preprint arXiv:1409.0473.
-
- Berman H.M. et al. (2002) The protein data bank. Acta Crystallogr. D Biol. Crystallogr., 58, 899–907. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
