

Gene-level quantitative trait mapping in Caenorhabditis elegans
- PMID: 33693602
- PMCID: PMC8022935
- DOI: 10.1093/g3journal/jkaa061
Gene-level quantitative trait mapping in Caenorhabditis elegans
Abstract
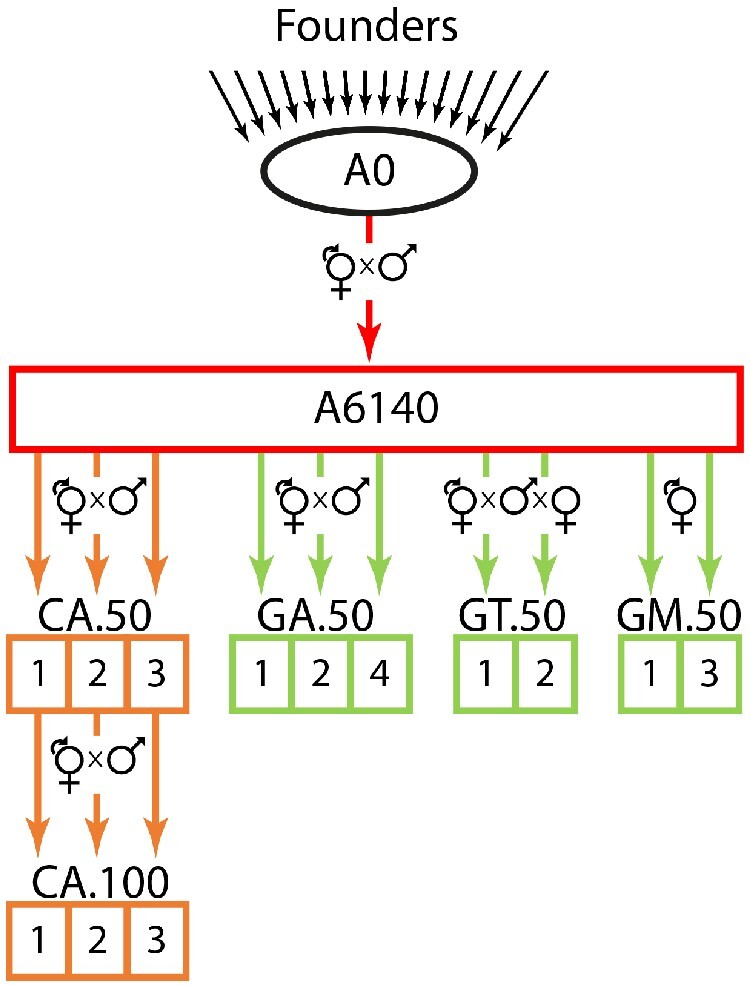
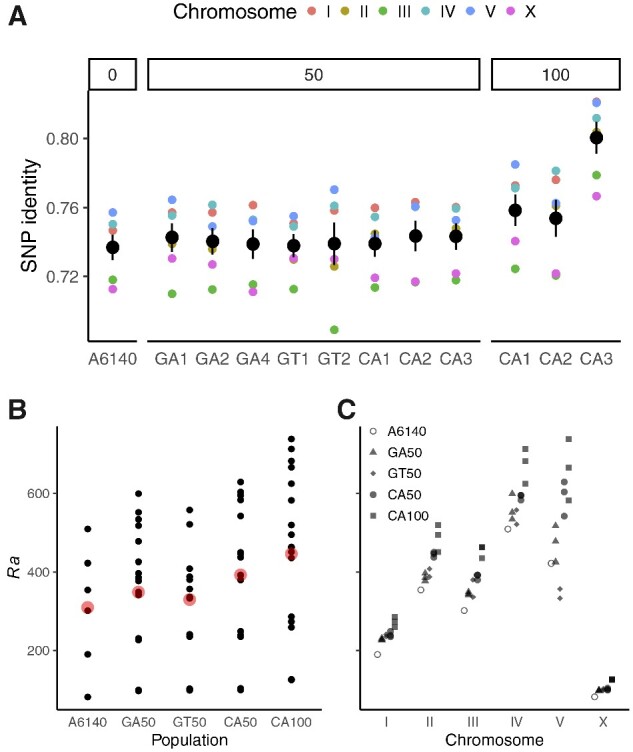
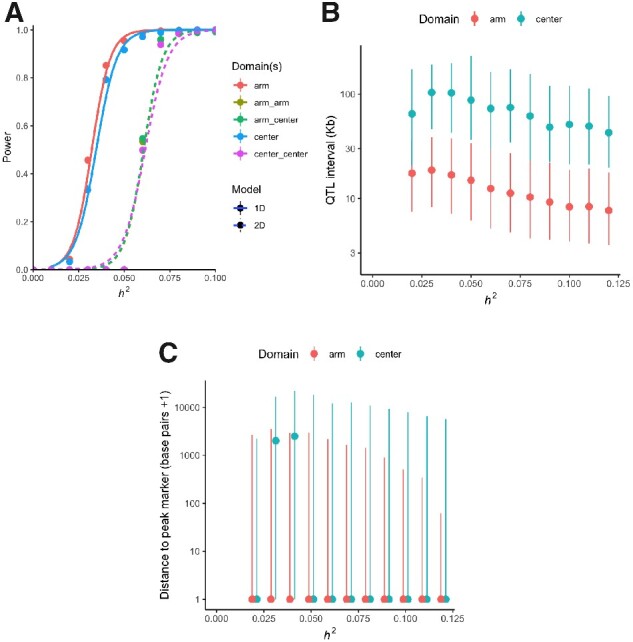
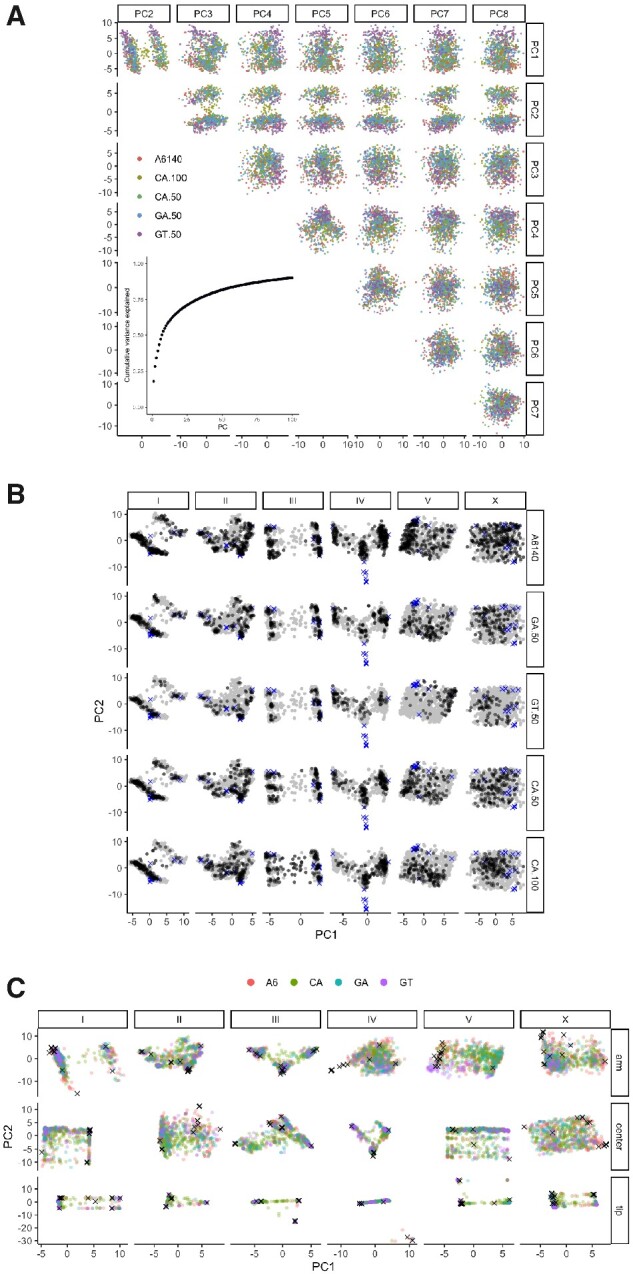
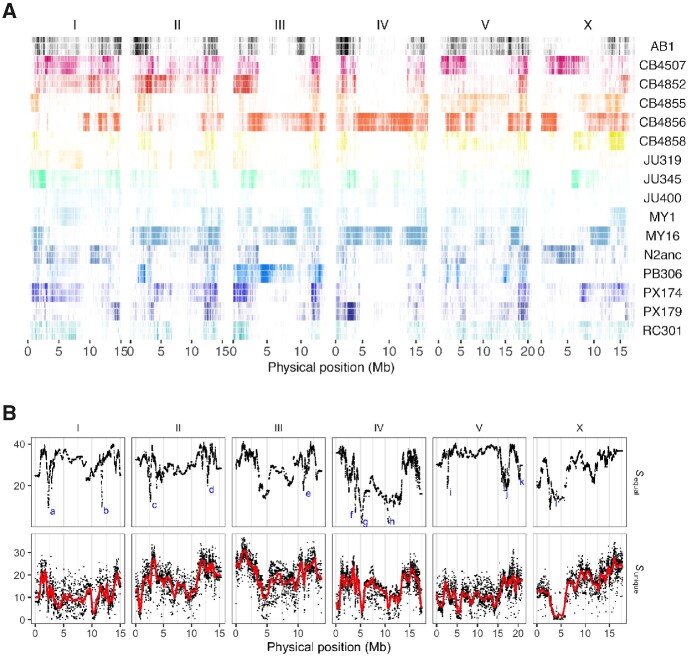
The Caenorhabditis elegans multiparental experimental evolution (CeMEE) panel is a collection of genome-sequenced, cryopreserved recombinant inbred lines useful for mapping the evolution and genetic basis of quantitative traits. We have expanded the resource with new lines and new populations, and here report the genotype and haplotype composition of CeMEE version 2, including a large set of putative de novo mutations, and updated additive and epistatic mapping simulations. Additive quantitative trait loci explaining 4% of trait variance are detected with >80% power, and the median detection interval approaches single-gene resolution on the highly recombinant chromosome arms. Although CeMEE populations are derived from a long-term evolution experiment, genetic structure is dominated by variation present in the ancestral population.
Keywords: MPP; Multiparent Advanced Generation Inter-Cross (MAGIC); Multiparental Populations; QTL; complex trait; experimental evolution; genetic architecture; quantitative trait.
© The Author(s) 2021. Published by Oxford University Press on behalf of Genetics Society of America.
Figures

References
-
- Barton NH, Keightley PD. 2002. Multifactorial genetics: understanding quantitative genetic variation. Nat Rev Genet. 3:11–21. - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources