

Lipid presentation by the protein C receptor links coagulation with autoimmunity
- PMID: 33707237
- PMCID: PMC9014225
- DOI: 10.1126/science.abc0956
Lipid presentation by the protein C receptor links coagulation with autoimmunity
Abstract
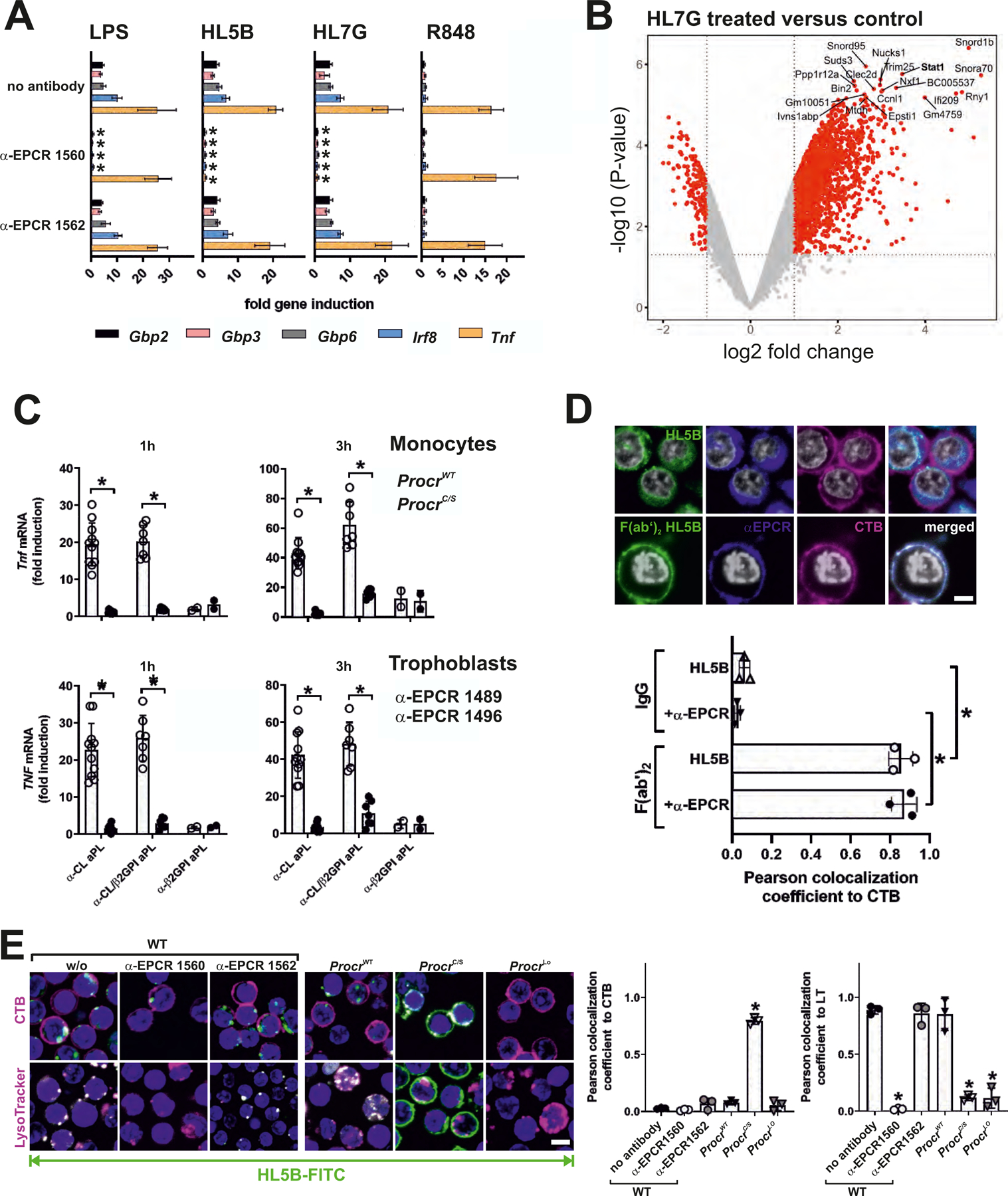
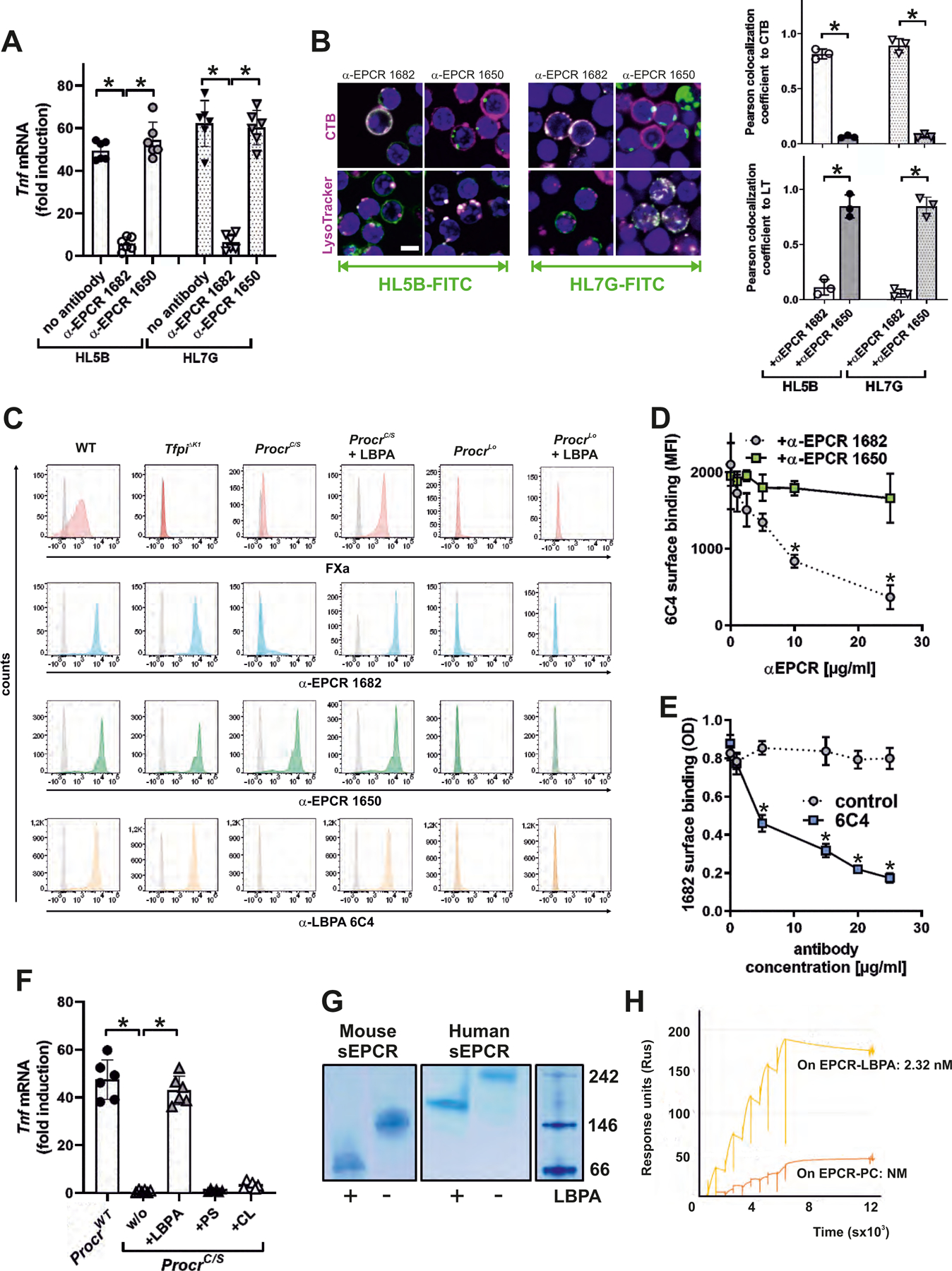
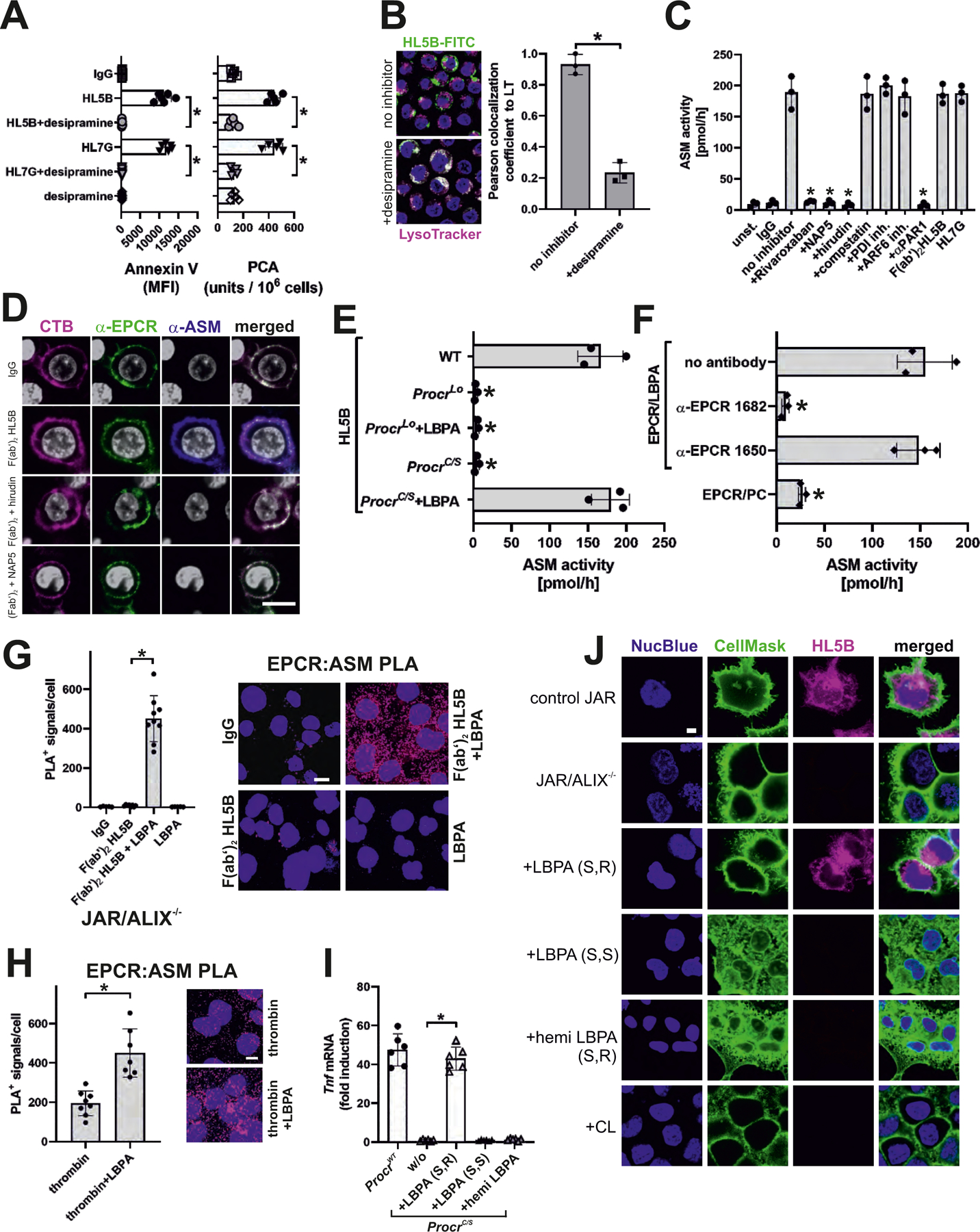
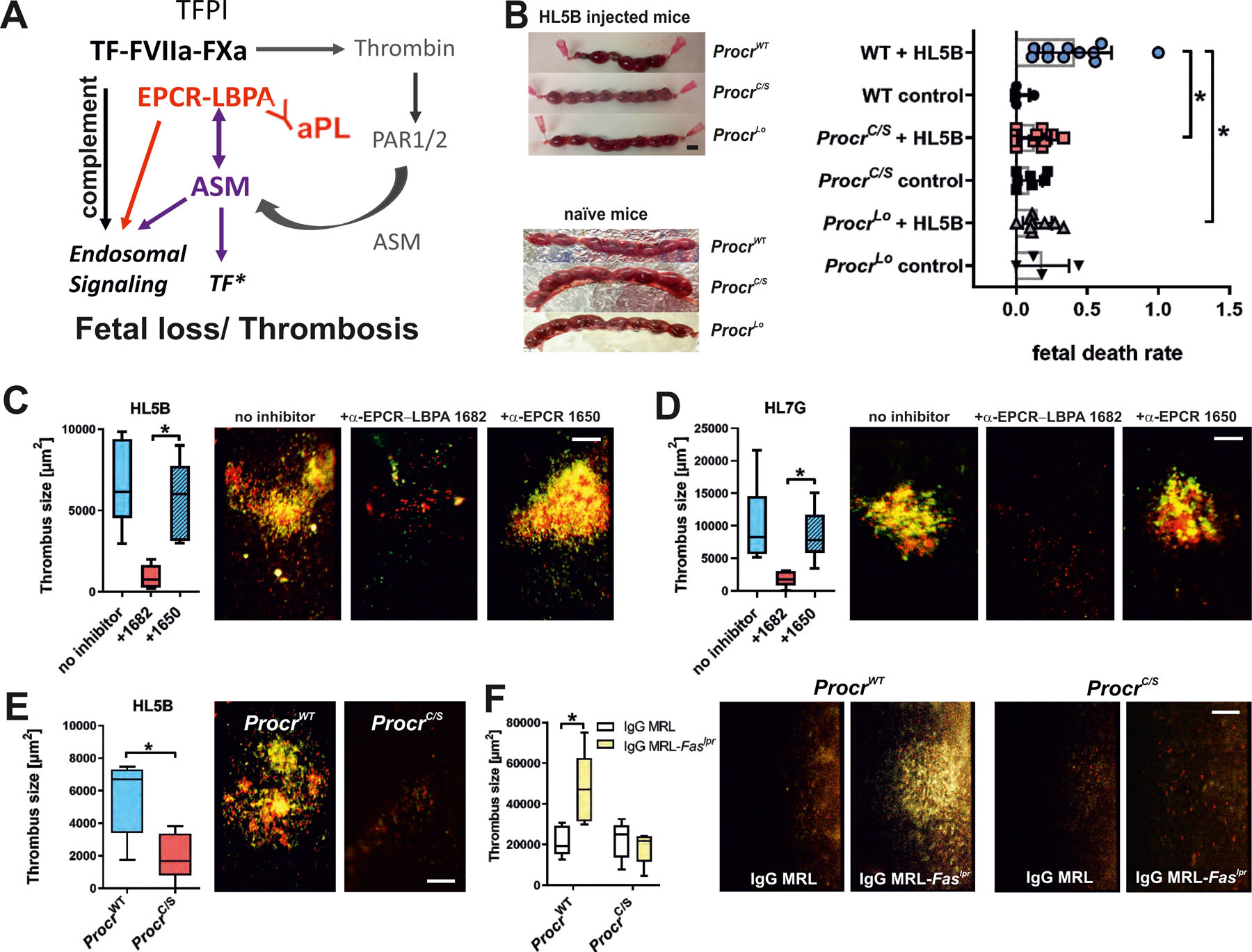
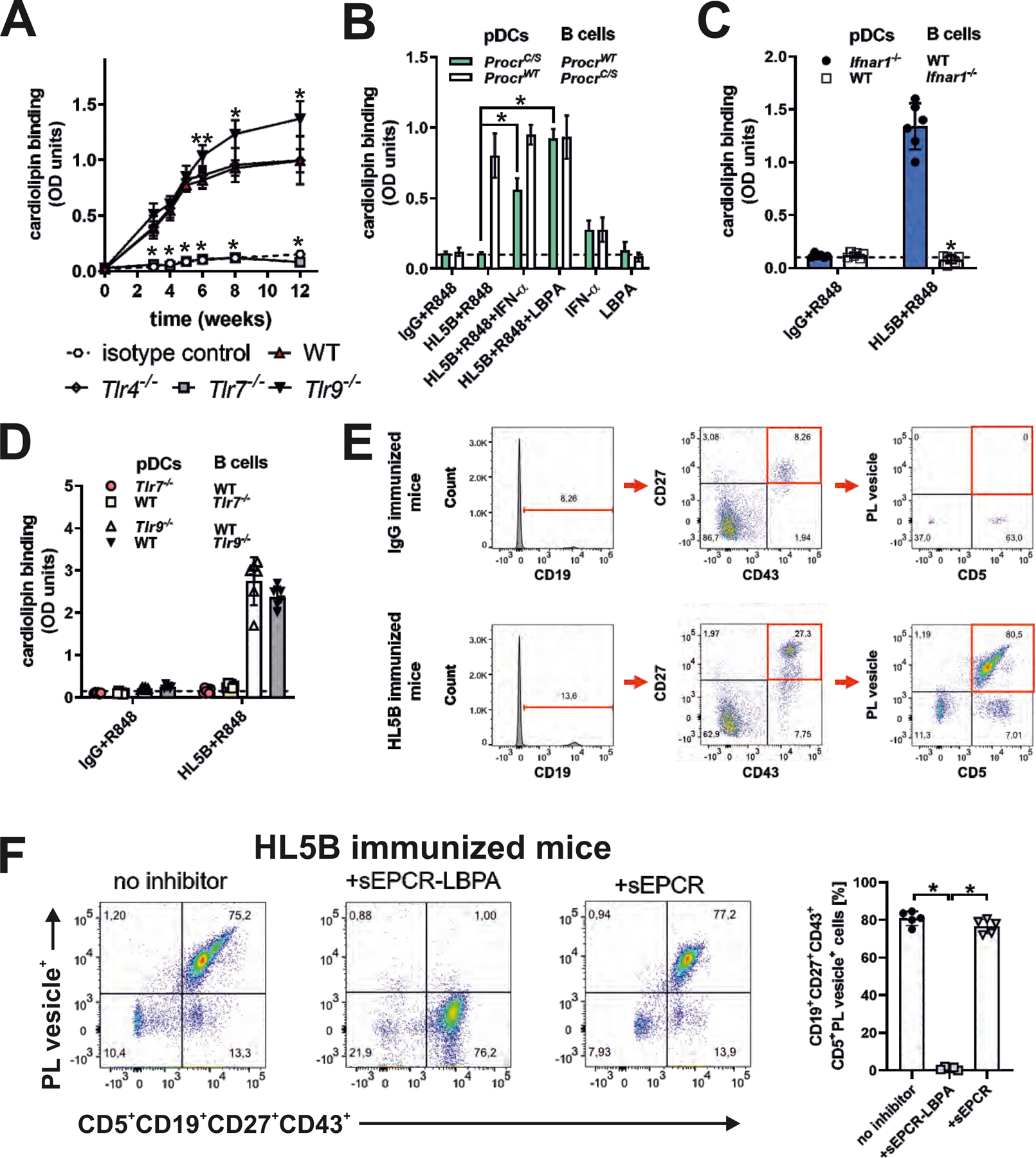
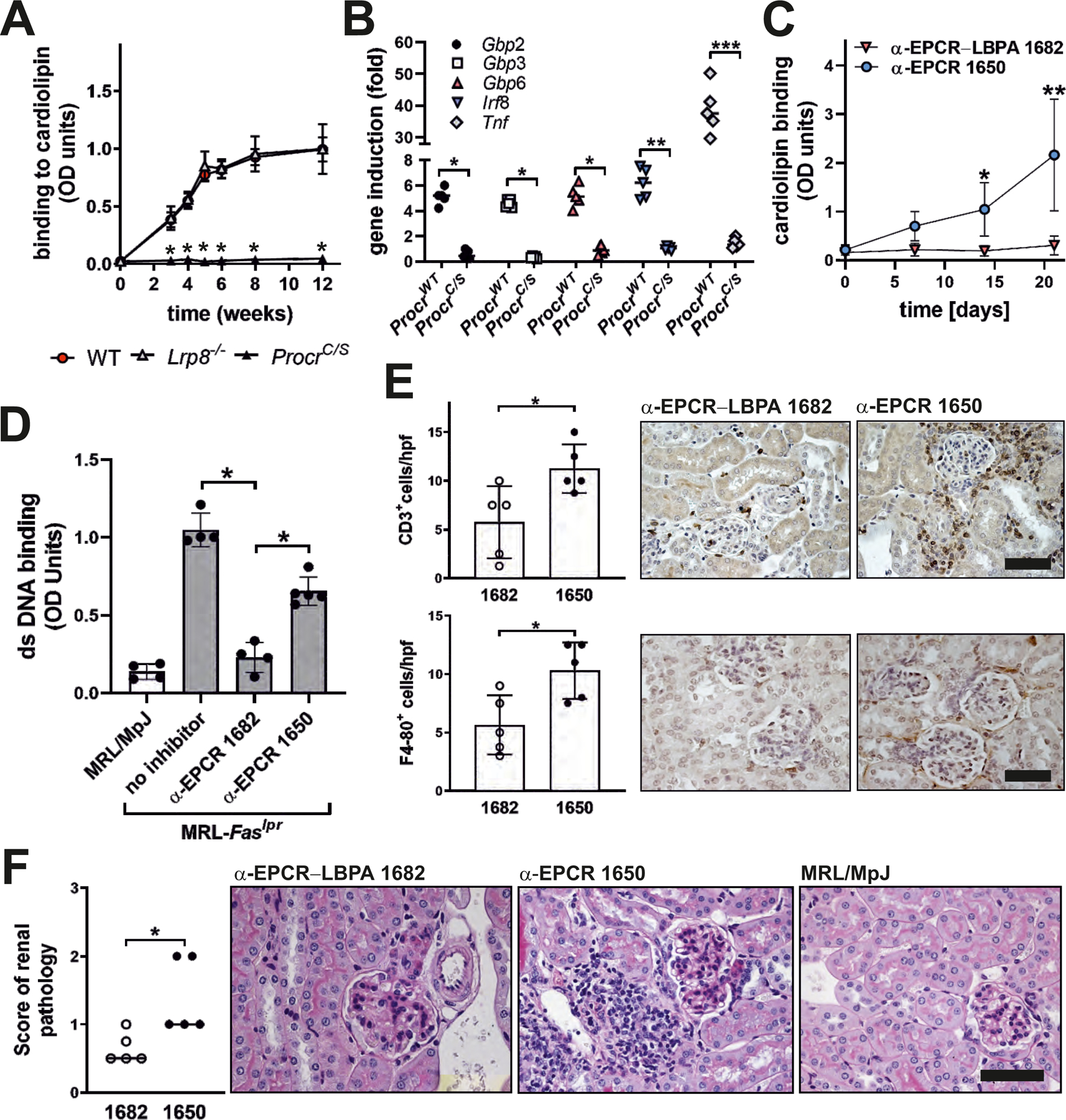
Antiphospholipid antibodies (aPLs) cause severe autoimmune disease characterized by vascular pathologies and pregnancy complications. Here, we identify endosomal lysobisphosphatidic acid (LBPA) presented by the CD1d-like endothelial protein C receptor (EPCR) as a pathogenic cell surface antigen recognized by aPLs for induction of thrombosis and endosomal inflammatory signaling. The engagement of aPLs with EPCR-LBPA expressed on innate immune cells sustains interferon- and toll-like receptor 7-dependent B1a cell expansion and autoantibody production. Specific pharmacological interruption of EPCR-LBPA signaling attenuates major aPL-elicited pathologies and the development of autoimmunity in a mouse model of systemic lupus erythematosus. Thus, aPLs recognize a single cell surface lipid-protein receptor complex to perpetuate a self-amplifying autoimmune signaling loop dependent on the cooperation with the innate immune complement and coagulation pathways.
Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Figures
Comment in
-
Linking clotting and autoimmunity.Science. 2021 Mar 12;371(6534):1100-1101. doi: 10.1126/science.abg6449. Science. 2021. PMID: 33707252 Free PMC article.
-
aPL target links coagulation and autoimmunity.Nat Rev Rheumatol. 2021 May;17(5):250. doi: 10.1038/s41584-021-00613-2. Nat Rev Rheumatol. 2021. PMID: 33828261 No abstract available.
-
The pivotal role of endothelial protein C receptor for antiphospholipid antibody-mediated pathologies.Rheumatology (Oxford). 2022 Mar 2;61(3):883-885. doi: 10.1093/rheumatology/keab620. Rheumatology (Oxford). 2022. PMID: 34324656 No abstract available.
References
-
- Giannakopoulos B, Krilis SA, The pathogenesis of the antiphospholipid syndrome. N. Engl. J Med 368, 1033–1044 (2013). - PubMed
-
- Garcia D, Erkan D, Diagnosis and Management of the Antiphospholipid Syndrome. N Engl J Med 378, 2010–2021 (2018). - PubMed
-
- Lieby P et al., The clonal analysis of anticardiolipin antibodies in a single patient with primary antiphospholipid syndrome reveals an extreme antibody heterogeneity. Blood 97, 3820–3828 (2001). - PubMed
-
- Prinz N, Hauser F, Lorenz M, Lackner KJ, von Landenberg P, Structural and functional characterization of a human IgG monoclonal antiphospholipid antibody. Immunobiology 216, 145–151 (2011). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
