

Enteric reabsorption processes and their impact on drug pharmacokinetics
- PMID: 33707635
- PMCID: PMC7952424
- DOI: 10.1038/s41598-021-85174-w
Enteric reabsorption processes and their impact on drug pharmacokinetics
Abstract
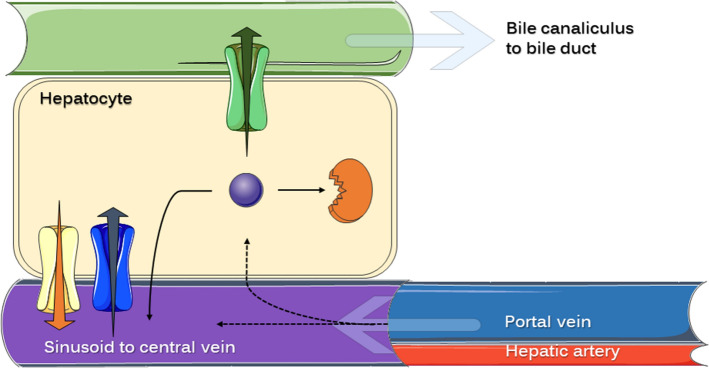
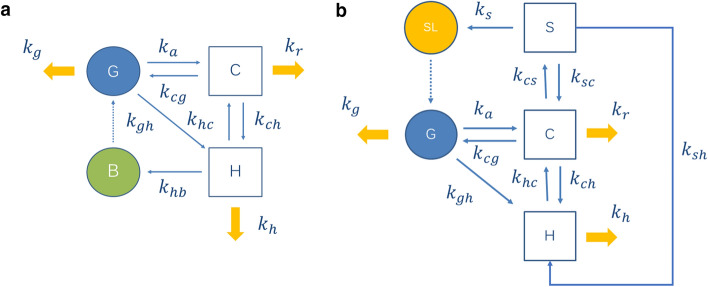
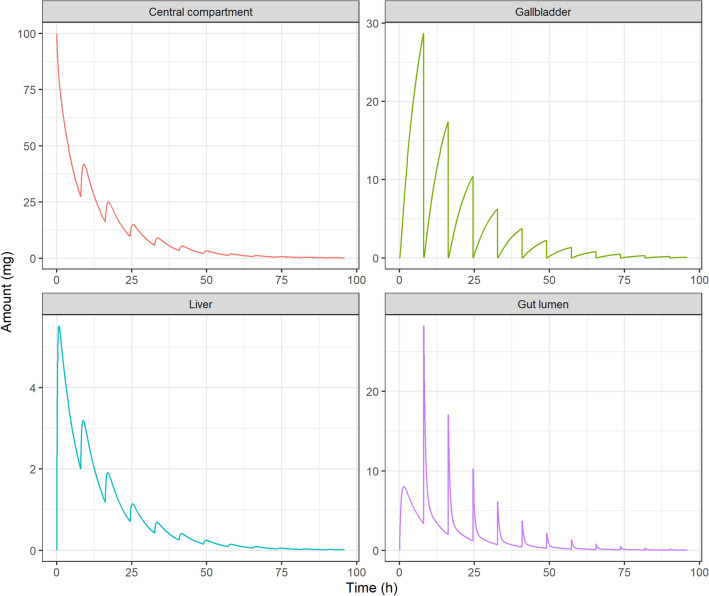
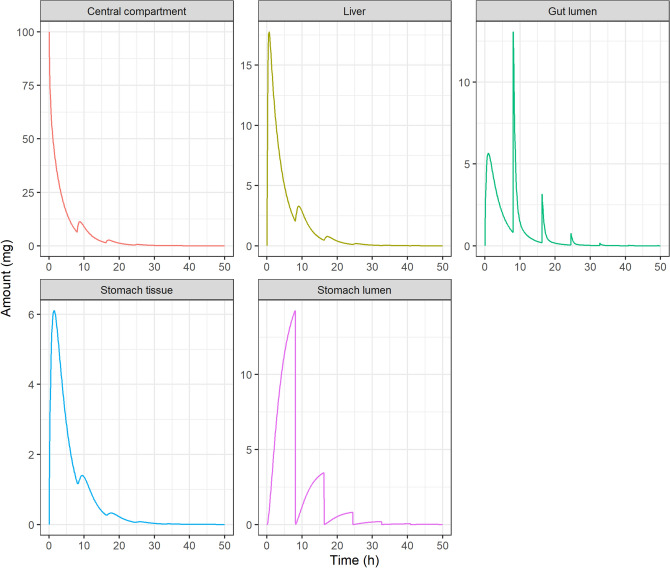
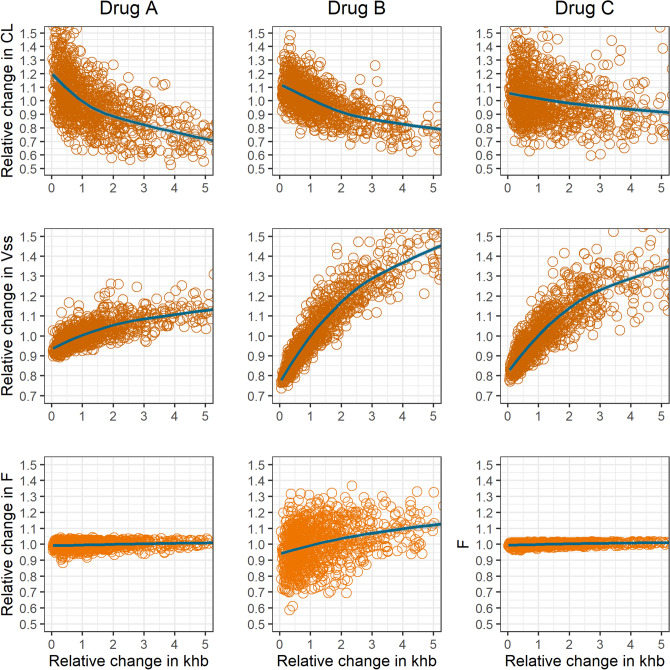
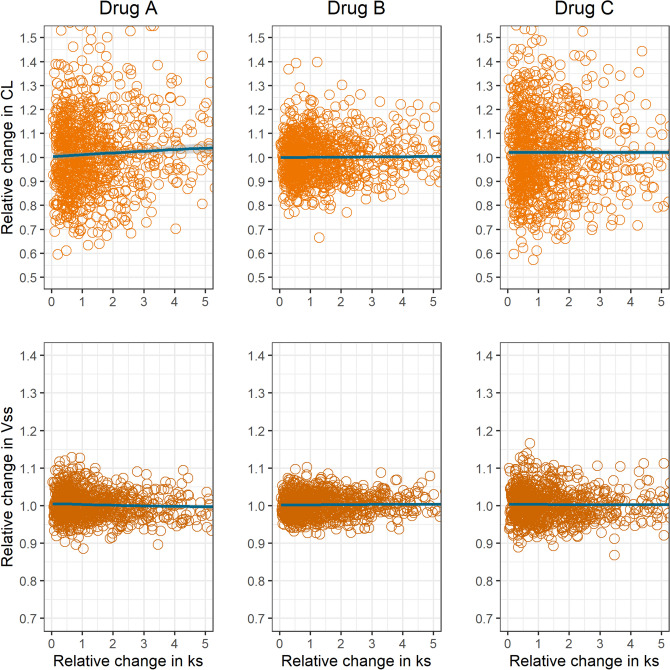
Enteric reabsorption occurs when a drug is secreted into the intestinal lumen and reabsorbed into the systemic circulation. This distribution process is evidenced by multiple peaks in pharmacokinetic profiles. Commonly, hepatobiliary drug secretion is assumed to be the underlying mechanism (enterohepatic reabsorption, EHR), neglecting other possible mechanisms such as gastric secretion (enterogastric reabsorption, EGR). In addition, the impact of drug reabsorption on systemic clearance, volume of distribution and bioavailability has been a subject of long-standing discussions. In this work, we propose semi-mechanistic pharmacokinetic models to reflect EHR and EGR and compare their respective impact on primary pharmacokinetic parameters. A simulation-based analysis was carried out considering three drug types with the potential for reabsorption, classified according to their primary route of elimination and their hepatic extraction: (A) hepatic metabolism-low extraction; (B) hepatic metabolism-intermediate/high extraction; (C) renal excretion. Results show that an increase in EHR can significantly reduce the clearance of drugs A and B, increase bioavailability of B drugs, and increase the volume of distribution for all drugs. Conversely, EGR had negligible impact in all pharmacokinetic parameters. Findings provide background to explain and forecast the role that this process can play in pharmacokinetic variability, including drug-drug interactions and disease states.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Okour M, Brundage RC. Modeling enterohepatic circulation. Curr. Pharmacol. Rep. 2017 doi: 10.1007/s40495-017-0096-z. - DOI
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
