

Extracellular Tuning of Mitochondrial Respiration Leads to Aortic Aneurysm
- PMID: 33709773
- PMCID: PMC8140666
- DOI: 10.1161/CIRCULATIONAHA.120.051171
Extracellular Tuning of Mitochondrial Respiration Leads to Aortic Aneurysm
Abstract
Background: Marfan syndrome (MFS) is an autosomal dominant disorder of the connective tissue caused by mutations in the FBN1 (fibrillin-1) gene encoding a large glycoprotein in the extracellular matrix called fibrillin-1. The major complication of this connective disorder is the risk to develop thoracic aortic aneurysm. To date, no effective pharmacologic therapies have been identified for the management of thoracic aortic disease and the only options capable of preventing aneurysm rupture are endovascular repair or open surgery. Here, we have studied the role of mitochondrial dysfunction in the progression of thoracic aortic aneurysm and mitochondrial boosting strategies as a potential treatment to managing aortic aneurysms.
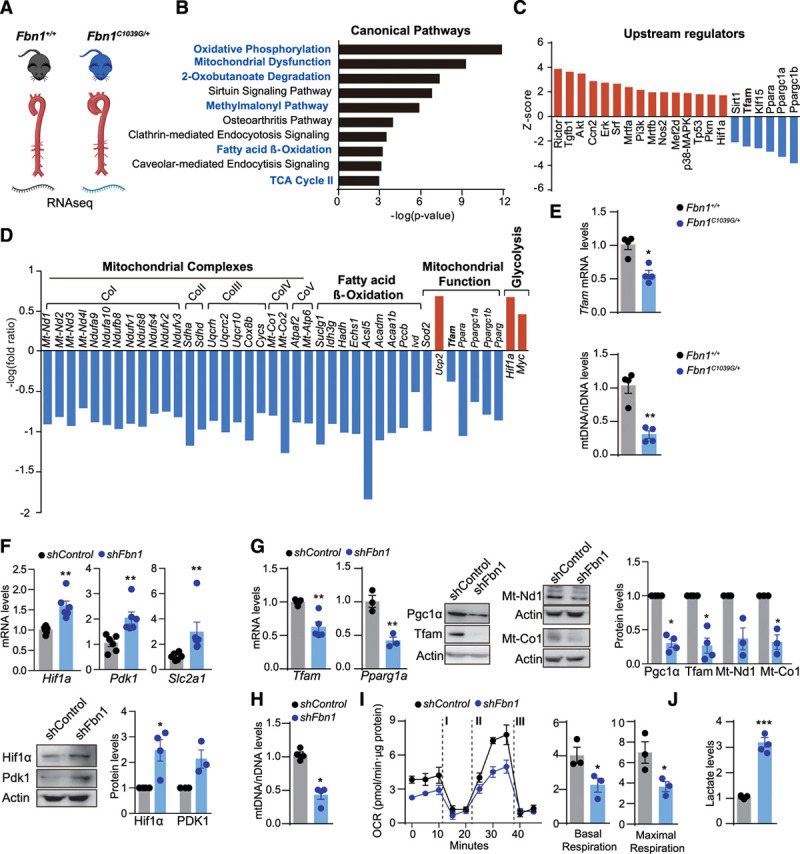
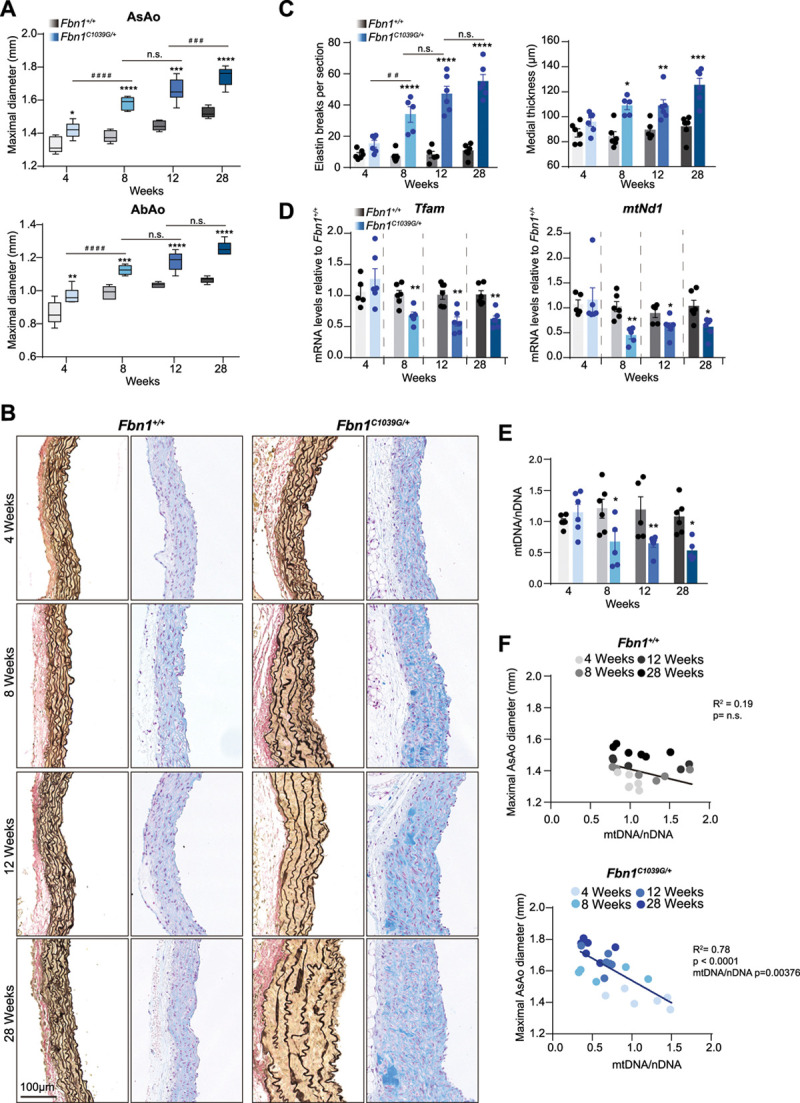
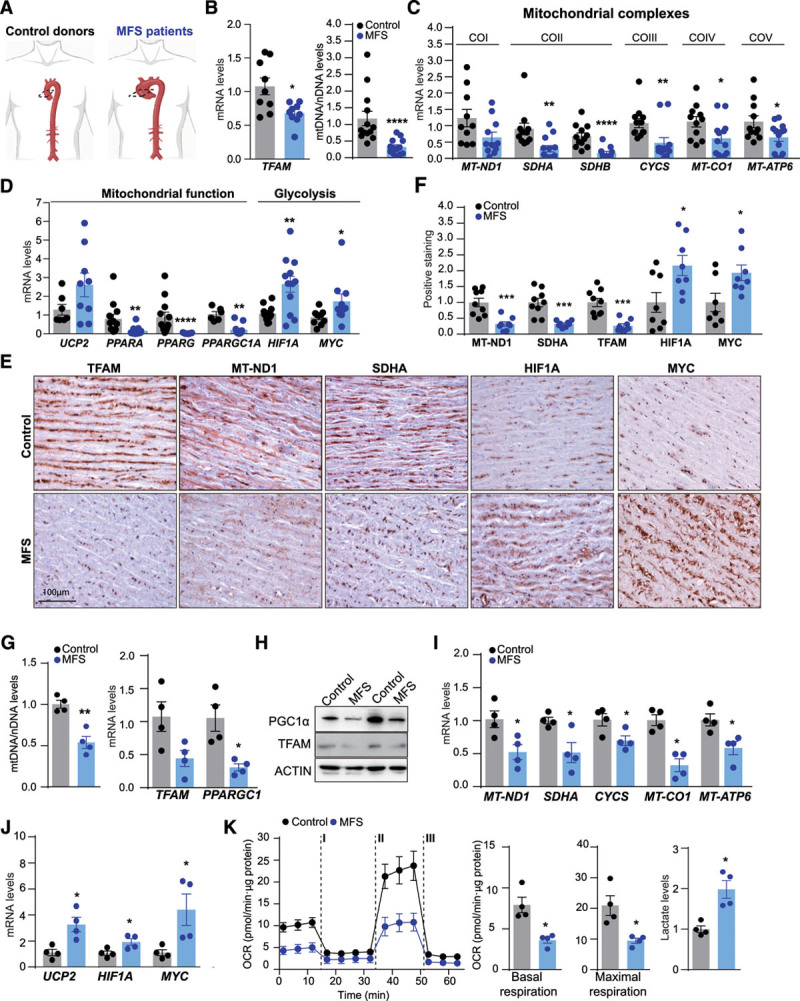
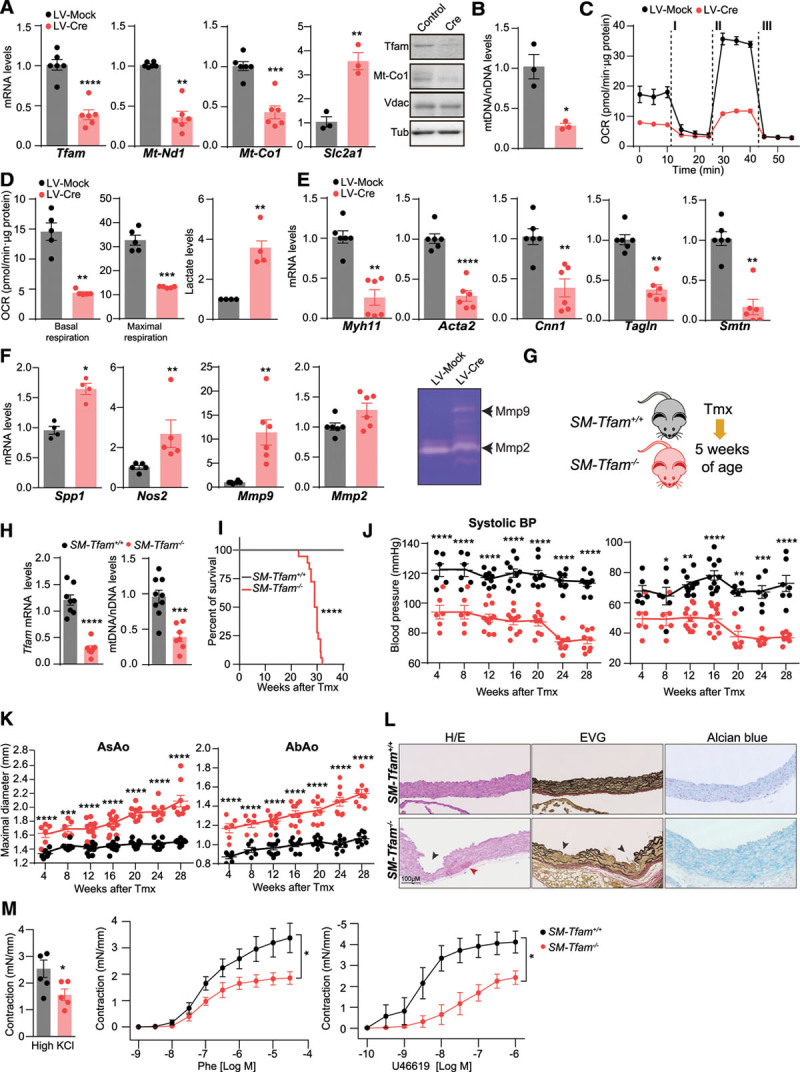
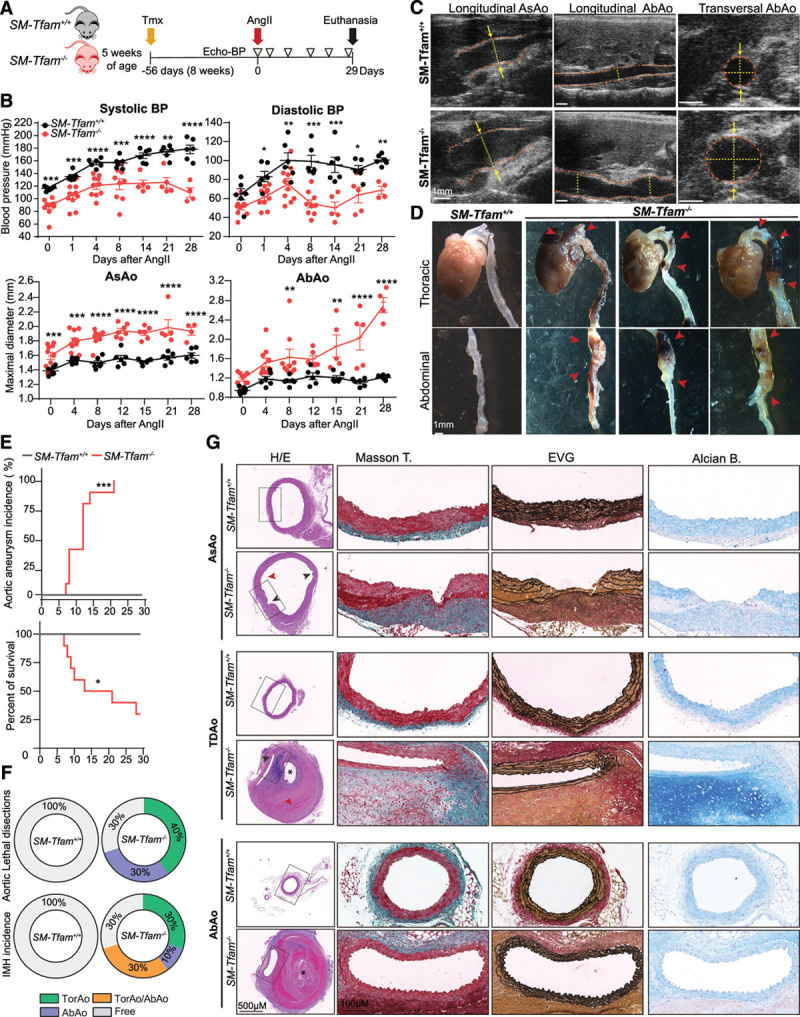
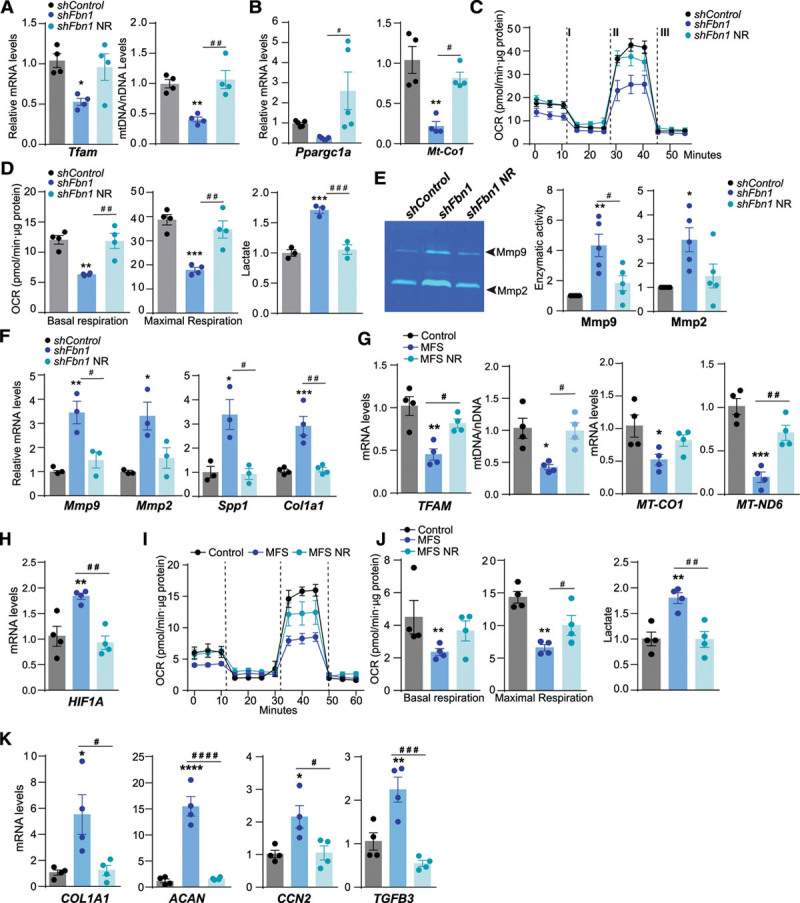
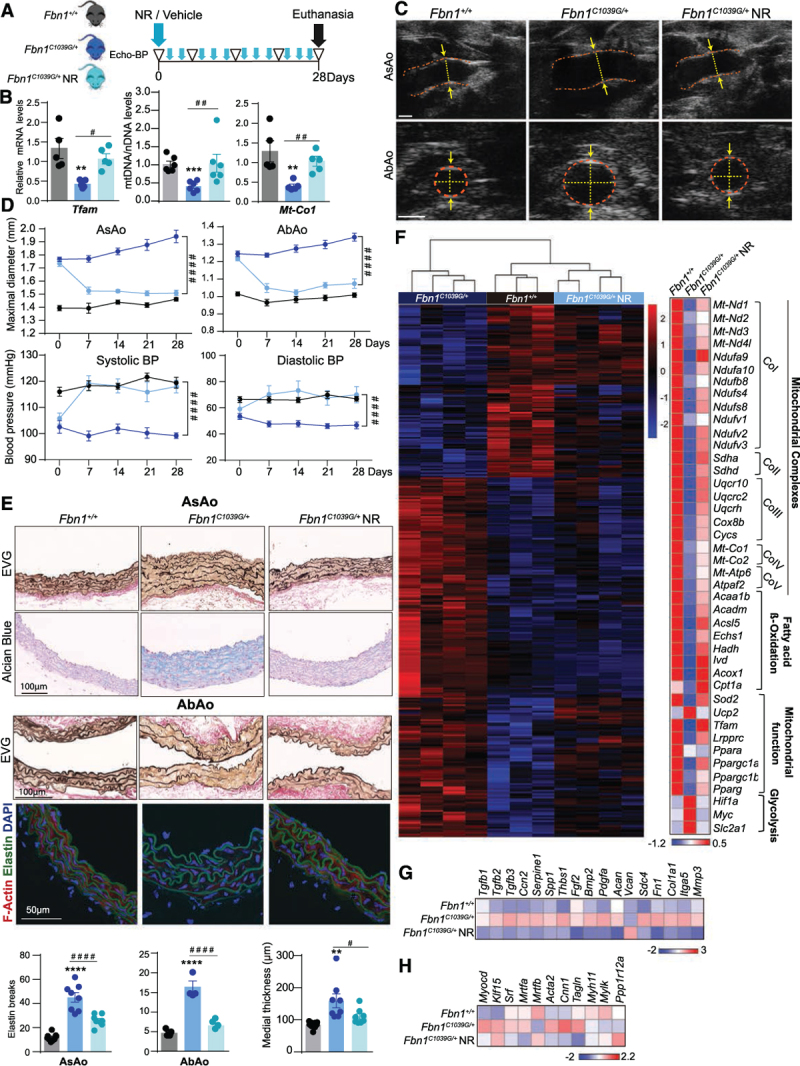
Methods: Combining transcriptomics and metabolic analysis of aortas from an MFS mouse model (Fbn1c1039g/+) and MFS patients, we have identified mitochondrial dysfunction alongside with mtDNA depletion as a new hallmark of aortic aneurysm disease in MFS. To demonstrate the importance of mitochondrial decline in the development of aneurysms, we generated a conditional mouse model with mitochondrial dysfunction specifically in vascular smooth muscle cells (VSMC) by conditional depleting Tfam (mitochondrial transcription factor A; Myh11-CreERT2Tfamflox/flox mice). We used a mouse model of MFS to test for drugs that can revert aortic disease by enhancing Tfam levels and mitochondrial respiration.
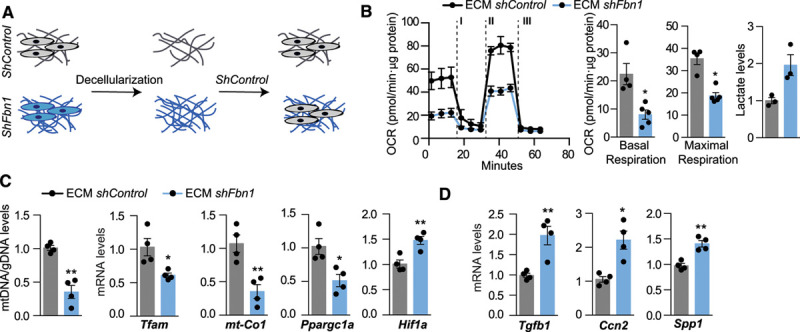
Results: The main canonical pathways highlighted in the transcriptomic analysis in aortas from Fbn1c1039g/+ mice were those related to metabolic function, such as mitochondrial dysfunction. Mitochondrial complexes, whose transcription depends on Tfam and mitochondrial DNA content, were reduced in aortas from young Fbn1c1039g/+ mice. In vitro experiments in Fbn1-silenced VSMCs presented increased lactate production and decreased oxygen consumption. Similar results were found in MFS patients. VSMCs seeded in matrices produced by Fbn1-deficient VSMCs undergo mitochondrial dysfunction. Conditional Tfam-deficient VSMC mice lose their contractile capacity, showed aortic aneurysms, and died prematurely. Restoring mitochondrial metabolism with the NAD precursor nicotinamide riboside rapidly reverses aortic aneurysm in Fbn1c1039g/+ mice.
Conclusions: Mitochondrial function of VSMCs is controlled by the extracellular matrix and drives the development of aortic aneurysm in Marfan syndrome. Targeting vascular metabolism is a new available therapeutic strategy for managing aortic aneurysms associated with genetic disorders.
Keywords: DNA, mitochondrial; Marfan syndrome; aortic aneurysm; extracellular matrix; genetic diseases, inborn; glycolysis; muscle, smooth, vascular.
Figures
References
-
- Gillis E, Van Laer L, Loeys BL. Genetics of thoracic aortic aneurysm: at the crossroad of transforming growth factor-β signaling and vascular smooth muscle cell contractility. Circ Res. 2013;113:327–340. doi: 10.1161/CIRCRESAHA.113.300675 - PubMed
-
- Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ, Curristin SM. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0 - PubMed
-
- Keane MG, Pyeritz RE. Medical management of Marfan syndrome. Circulation. 2008;117:2802–2813. doi: 10.1161/CIRCULATIONAHA.107.693523 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
