

Protein sequence design by conformational landscape optimization
- PMID: 33712545
- PMCID: PMC7980421
- DOI: 10.1073/pnas.2017228118
Protein sequence design by conformational landscape optimization
Abstract
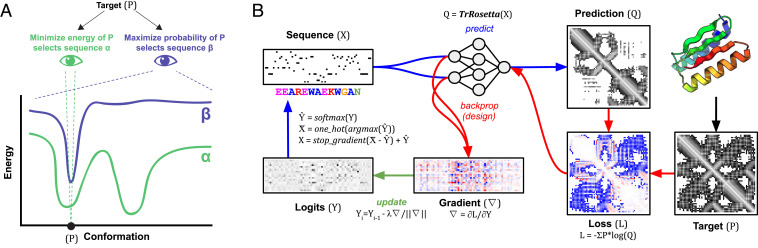
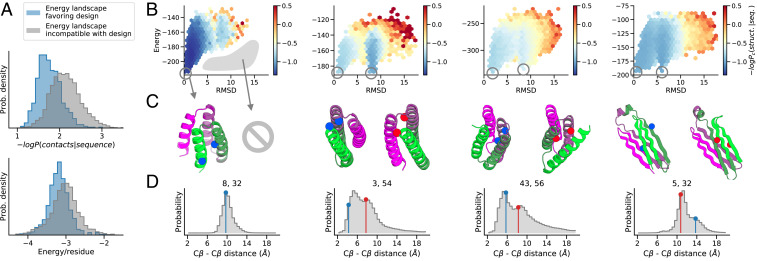
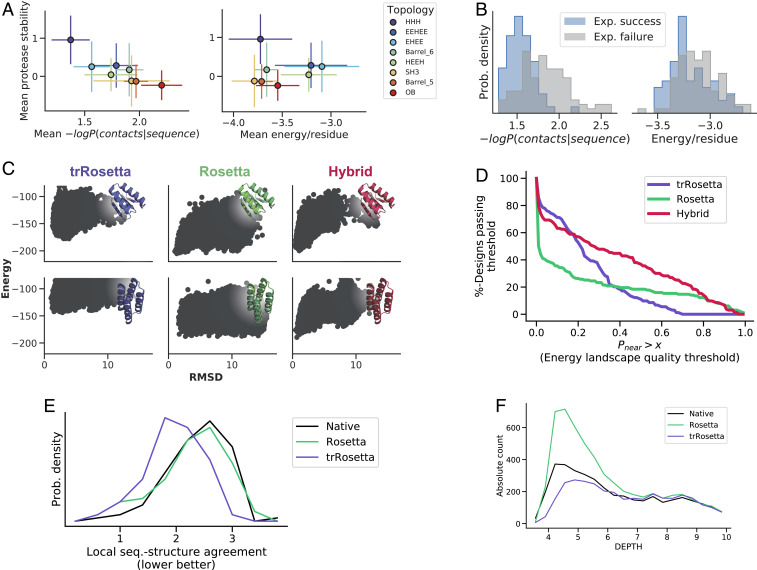
The protein design problem is to identify an amino acid sequence that folds to a desired structure. Given Anfinsen's thermodynamic hypothesis of folding, this can be recast as finding an amino acid sequence for which the desired structure is the lowest energy state. As this calculation involves not only all possible amino acid sequences but also, all possible structures, most current approaches focus instead on the more tractable problem of finding the lowest-energy amino acid sequence for the desired structure, often checking by protein structure prediction in a second step that the desired structure is indeed the lowest-energy conformation for the designed sequence, and typically discarding a large fraction of designed sequences for which this is not the case. Here, we show that by backpropagating gradients through the transform-restrained Rosetta (trRosetta) structure prediction network from the desired structure to the input amino acid sequence, we can directly optimize over all possible amino acid sequences and all possible structures in a single calculation. We find that trRosetta calculations, which consider the full conformational landscape, can be more effective than Rosetta single-point energy estimations in predicting folding and stability of de novo designed proteins. We compare sequence design by conformational landscape optimization with the standard energy-based sequence design methodology in Rosetta and show that the former can result in energy landscapes with fewer alternative energy minima. We show further that more funneled energy landscapes can be designed by combining the strengths of the two approaches: the low-resolution trRosetta model serves to disfavor alternative states, and the high-resolution Rosetta model serves to create a deep energy minimum at the design target structure.
Keywords: energy landscape; machine learning; protein design; sequence optimization; stability prediction.
Copyright © 2021 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
-
- Kuhlman B., et al., Design of a novel globular protein fold with atomic-level accuracy. Science 302, 1364–1368 (2003). - PubMed
-
- Dahiyat B. I., Mayo S. L., De novo protein design: Fully automated sequence selection. Science 278, 82–87 (1997). - PubMed
-
- Ingraham J., Garg V., Barzilay R., Jaakkola T., Generative models for graph-based protein design. NeurIPS Proc. 32, 15820–15831 (2019).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
