

Integration of Alzheimer's disease genetics and myeloid genomics identifies disease risk regulatory elements and genes
- PMID: 33712570
- PMCID: PMC7955030
- DOI: 10.1038/s41467-021-21823-y
Integration of Alzheimer's disease genetics and myeloid genomics identifies disease risk regulatory elements and genes
Abstract
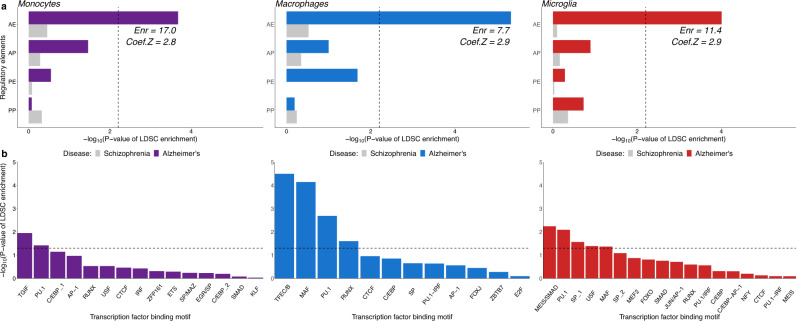
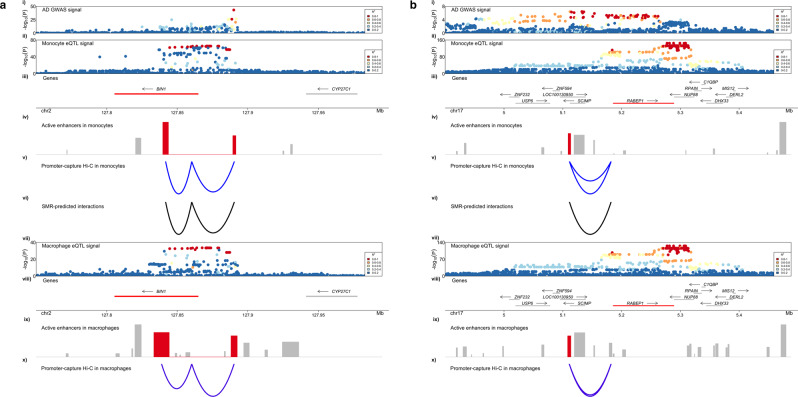
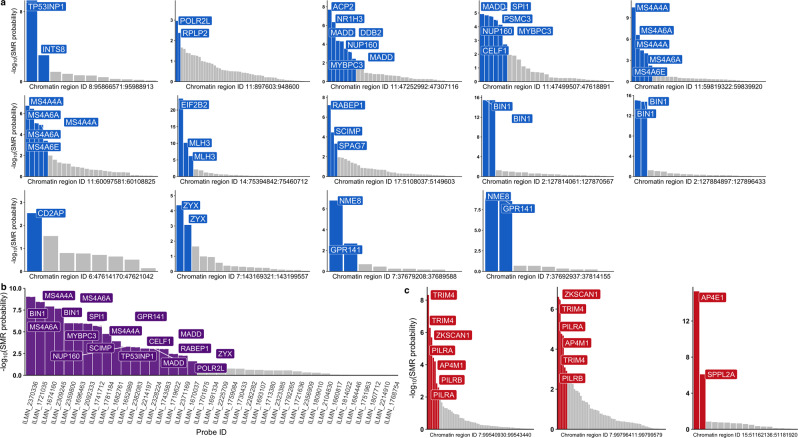
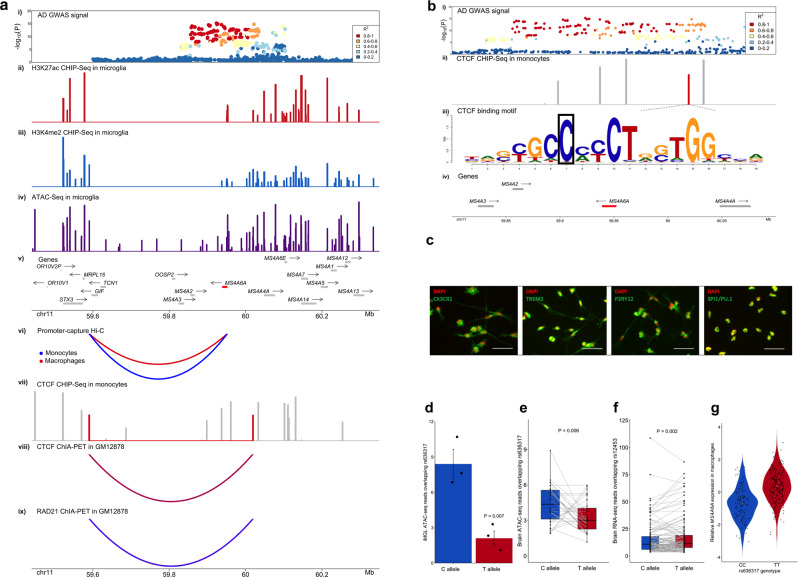
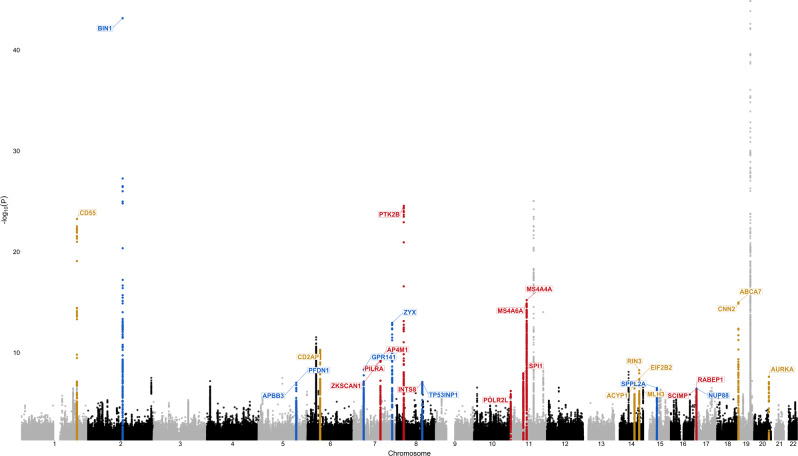
Genome-wide association studies (GWAS) have identified more than 40 loci associated with Alzheimer's disease (AD), but the causal variants, regulatory elements, genes and pathways remain largely unknown, impeding a mechanistic understanding of AD pathogenesis. Previously, we showed that AD risk alleles are enriched in myeloid-specific epigenomic annotations. Here, we show that they are specifically enriched in active enhancers of monocytes, macrophages and microglia. We integrated AD GWAS with myeloid epigenomic and transcriptomic datasets using analytical approaches to link myeloid enhancer activity to target gene expression regulation and AD risk modification. We identify AD risk enhancers and nominate candidate causal genes among their likely targets (including AP4E1, AP4M1, APBB3, BIN1, MS4A4A, MS4A6A, PILRA, RABEP1, SPI1, TP53INP1, and ZYX) in twenty loci. Fine-mapping of these enhancers nominates candidate functional variants that likely modify AD risk by regulating gene expression in myeloid cells. In the MS4A locus we identified a single candidate functional variant and validated it in human induced pluripotent stem cell (hiPSC)-derived microglia and brain. Taken together, this study integrates AD GWAS with multiple myeloid genomic datasets to investigate the mechanisms of AD risk alleles and nominates candidate functional variants, regulatory elements and genes that likely modulate disease susceptibility.
Conflict of interest statement
A.M.G. has consulted for Eisai, Biogen, Pfizer, AbbVie, Cognition Therapeutics and GSK. She also served on the SAB at Denali Therapeutics from 2015 to 2018. This work was funded by grants from the NIH: U01AG052411 (A.M.G.), RF1AG054011 (A.M.G.), U01AG058635 (A.M.G.), NIA K01AG062683 (J.TCW.), AG016573 (W.W.P.), F31 AG059337-01 (A.G.E.), R01AG050986 (P.R.), 1R01ES029212-01 (K.H.), R01HL125863 (J.L.M.B.), American Heart Association (J.L.M.B.), the Swedish Research Council (J.L.M.B.), Heart Lung Foundation (J.L.M.B.), and by Astra-Zeneca through ICMC, Karolinska Institutet (J.L.M.B.), The JPB Foundation, The Robert and Renee Belfer Foundation. E.M.A. and W.W.P. are named co-inventors of patent WO/2018/160496 related to the differentiation and use of human pluripotent stem cells and hematopoietic progenitors into microglia. K.H. receives financial compensation from Sema4 (an Icahn School of Medicine at Mount Sinai spin-off company). Sema4 is currently majority owned by the Icahn School of Medicine at Mount Sinai. The remaining authors declare no competing interests.
Figures
References
-
- Dementia statistics | Alzheimer’s Disease International. https://www.alz.co.uk/research/statistics.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
