

Global discovery of lupus genetic risk variant allelic enhancer activity
- PMID: 33712590
- PMCID: PMC7955039
- DOI: 10.1038/s41467-021-21854-5
Global discovery of lupus genetic risk variant allelic enhancer activity
Abstract
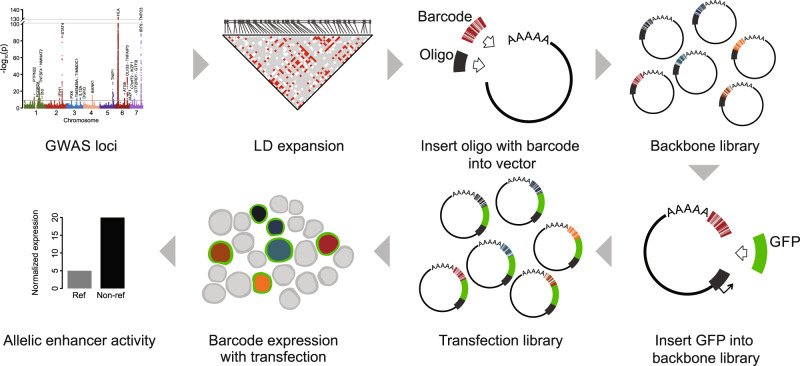
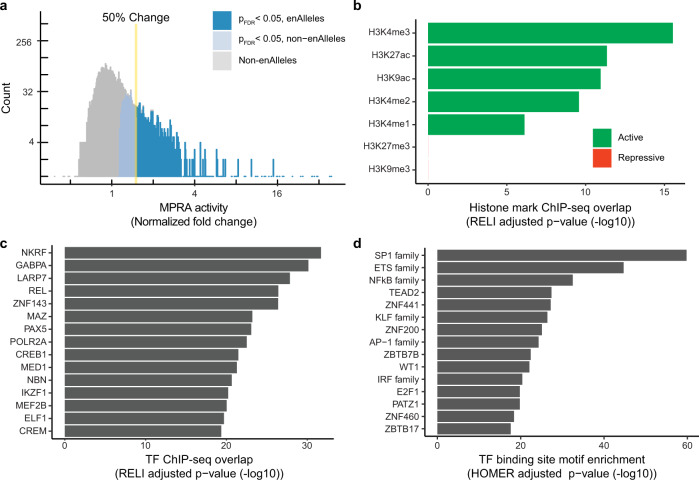
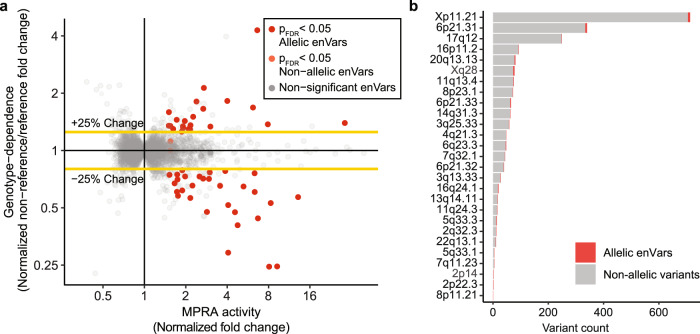
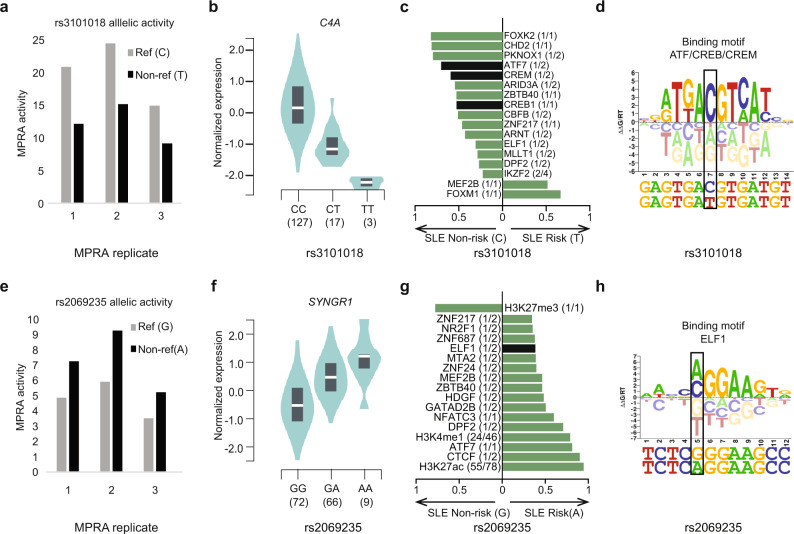
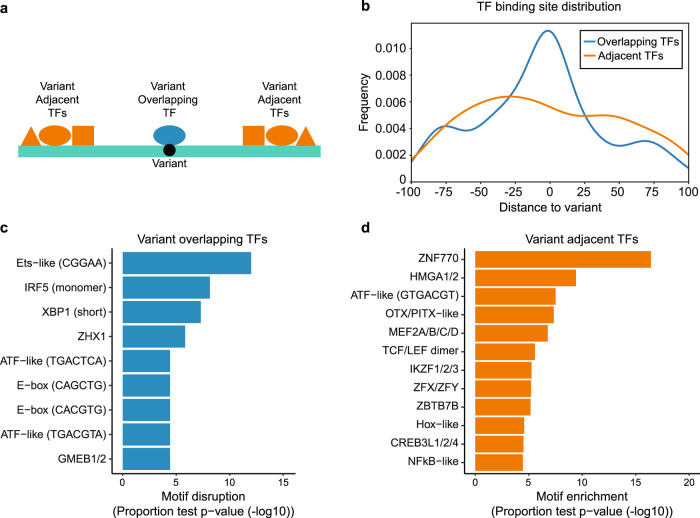
Genome-wide association studies of Systemic Lupus Erythematosus (SLE) nominate 3073 genetic variants at 91 risk loci. To systematically screen these variants for allelic transcriptional enhancer activity, we construct a massively parallel reporter assay (MPRA) library comprising 12,396 DNA oligonucleotides containing the genomic context around every allele of each SLE variant. Transfection into the Epstein-Barr virus-transformed B cell line GM12878 reveals 482 variants with enhancer activity, with 51 variants showing genotype-dependent (allelic) enhancer activity at 27 risk loci. Comparison of MPRA results in GM12878 and Jurkat T cell lines highlights shared and unique allelic transcriptional regulatory mechanisms at SLE risk loci. In-depth analysis of allelic transcription factor (TF) binding at and around allelic variants identifies one class of TFs whose DNA-binding motif tends to be directly altered by the risk variant and a second class of TFs that bind allelically without direct alteration of their motif by the variant. Collectively, our approach provides a blueprint for the discovery of allelic gene regulation at risk loci for any disease and offers insight into the transcriptional regulatory mechanisms underlying SLE.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- U01 AI130830/AI/NIAID NIH HHS/United States
- R01 NS099068/NS/NINDS NIH HHS/United States
- R01 DK107502/DK/NIDDK NIH HHS/United States
- R01 HG010730/HG/NHGRI NIH HHS/United States
- K22 AI153648/AI/NIAID NIH HHS/United States
- K99 HG009920/HG/NHGRI NIH HHS/United States
- R01 GM055479/GM/NIGMS NIH HHS/United States
- P30 AR070549/AR/NIAMS NIH HHS/United States
- R01 AI148276/AI/NIAID NIH HHS/United States
- I01 BX001834/BX/BLRD VA/United States
- F32 AI129249/AI/NIAID NIH HHS/United States
- R01 AR073228/AR/NIAMS NIH HHS/United States
- U01 HG011172/HG/NHGRI NIH HHS/United States
- P30 DK078392/DK/NIDDK NIH HHS/United States
- U01 AI150748/AI/NIAID NIH HHS/United States
- R01 AI024717/AI/NIAID NIH HHS/United States
- U01 HG008666/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
