

Single-cell transcriptional changes associated with drug tolerance and response to combination therapies in cancer
- PMID: 33712615
- PMCID: PMC7955121
- DOI: 10.1038/s41467-021-21884-z
Single-cell transcriptional changes associated with drug tolerance and response to combination therapies in cancer
Abstract
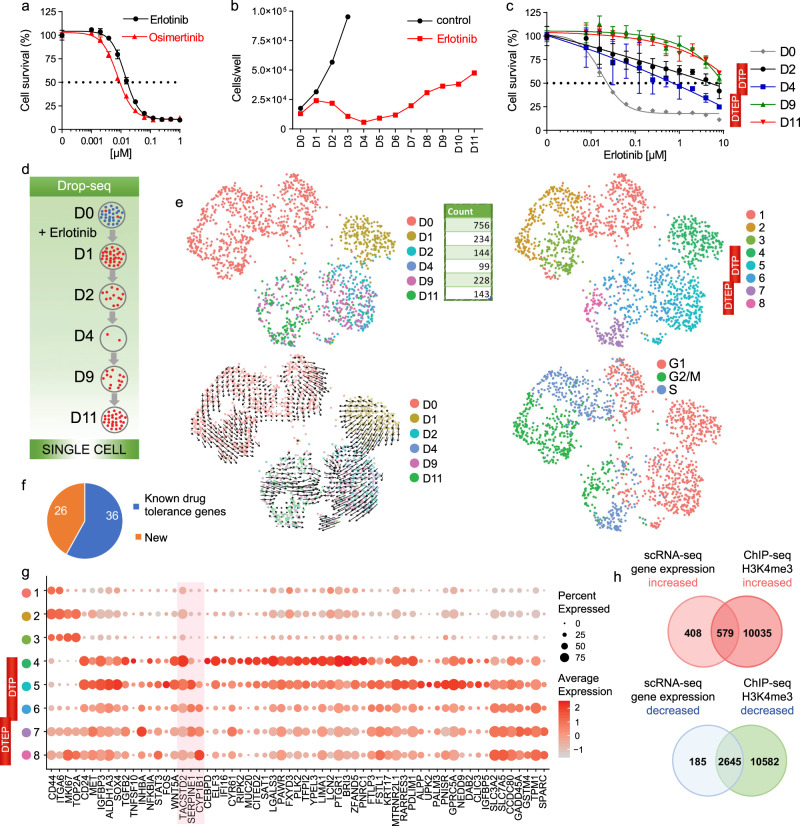
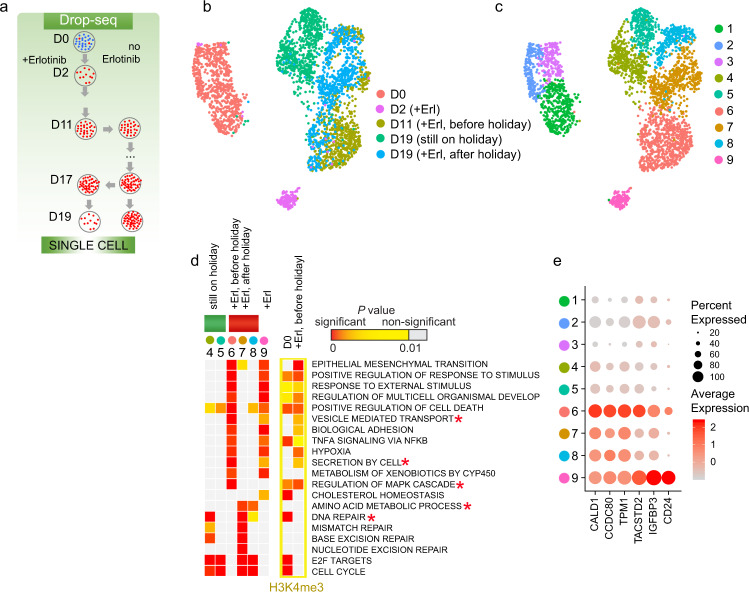
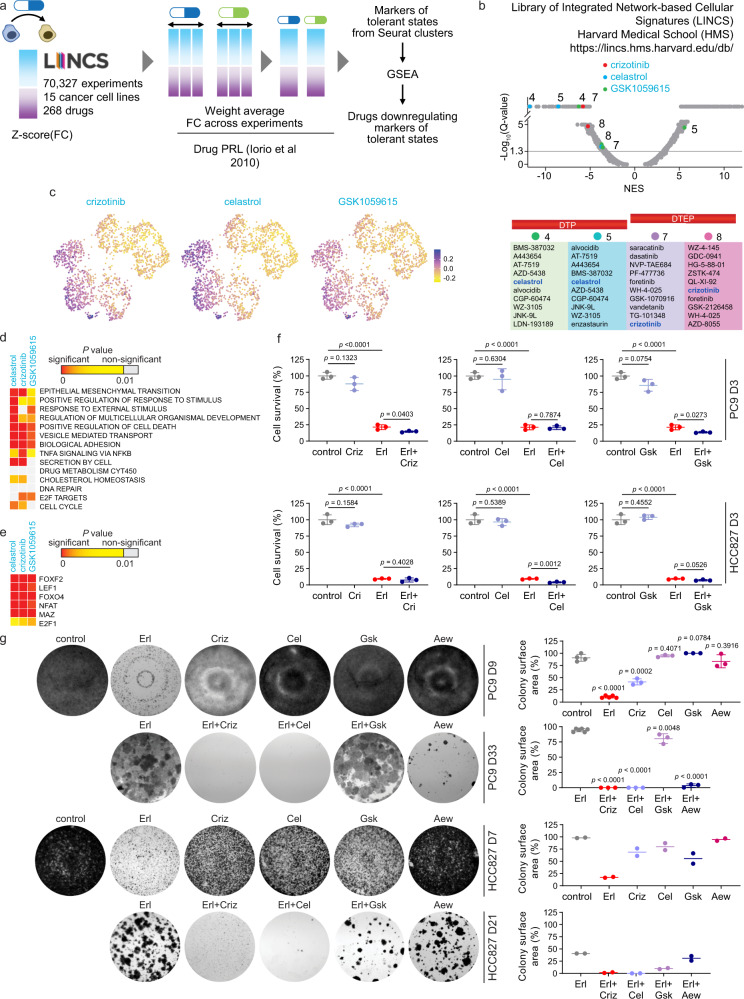
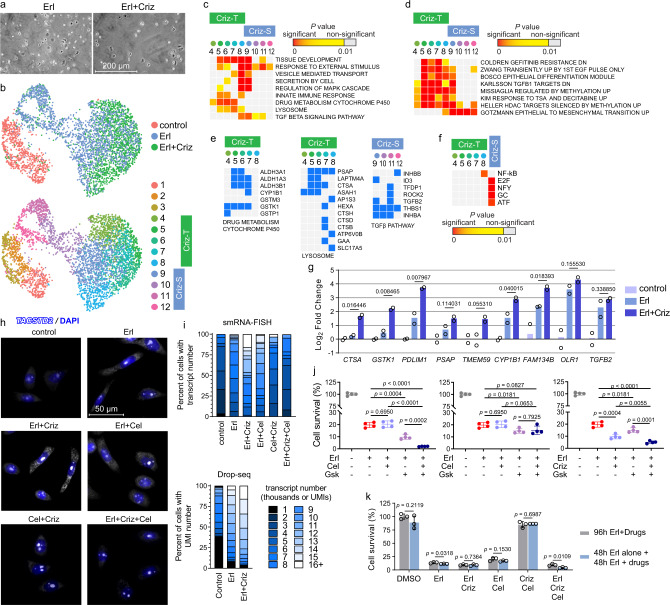
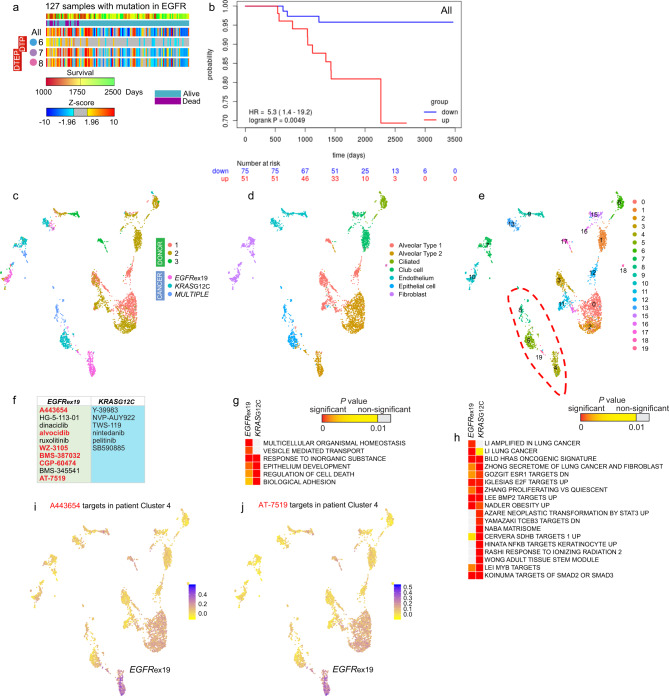
Tyrosine kinase inhibitors were found to be clinically effective for treatment of patients with certain subsets of cancers carrying somatic mutations in receptor tyrosine kinases. However, the duration of clinical response is often limited, and patients ultimately develop drug resistance. Here, we use single-cell RNA sequencing to demonstrate the existence of multiple cancer cell subpopulations within cell lines, xenograft tumors and patient tumors. These subpopulations exhibit epigenetic changes and differential therapeutic sensitivity. Recurrently overrepresented ontologies in genes that are differentially expressed between drug tolerant cell populations and drug sensitive cells include epithelial-to-mesenchymal transition, epithelium development, vesicle mediated transport, drug metabolism and cholesterol homeostasis. We show analysis of identified markers using the LINCS database to predict and functionally validate small molecules that target selected drug tolerant cell populations. In combination with EGFR inhibitors, crizotinib inhibits the emergence of a defined subset of EGFR inhibitor-tolerant clones. In this study, we describe the spectrum of changes associated with drug tolerance and inhibition of specific tolerant cell subpopulations with combination agents.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
-
- Amann J, et al. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res. 2005;65:226–235. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
