

The SARS-CoV-2 subgenome landscape and its novel regulatory features
- PMID: 33713597
- PMCID: PMC7927579
- DOI: 10.1016/j.molcel.2021.02.036
The SARS-CoV-2 subgenome landscape and its novel regulatory features
Abstract
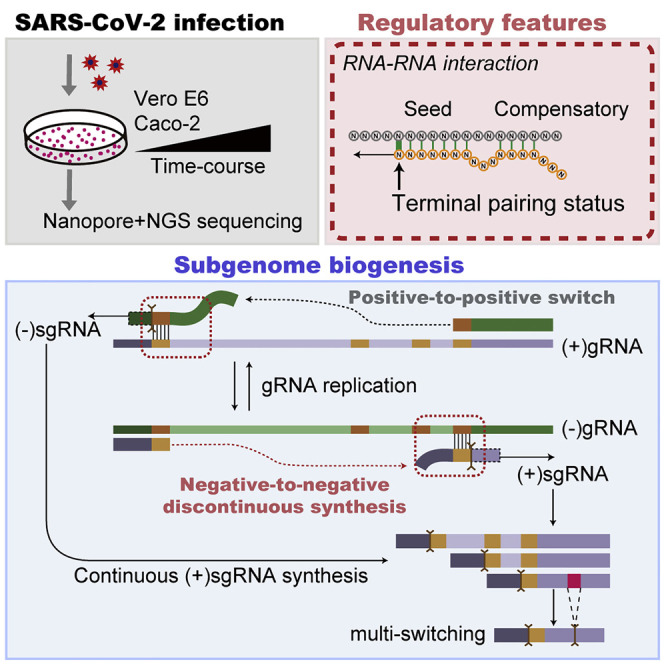
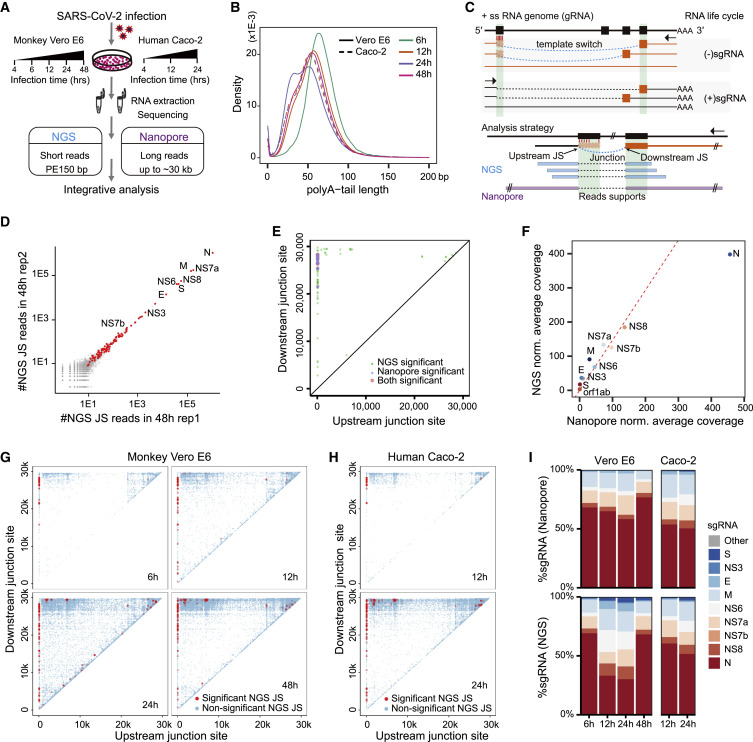
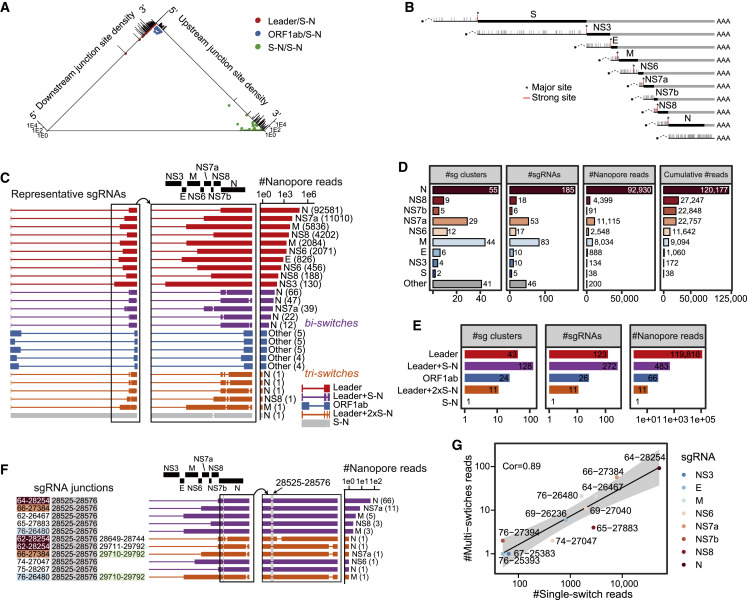
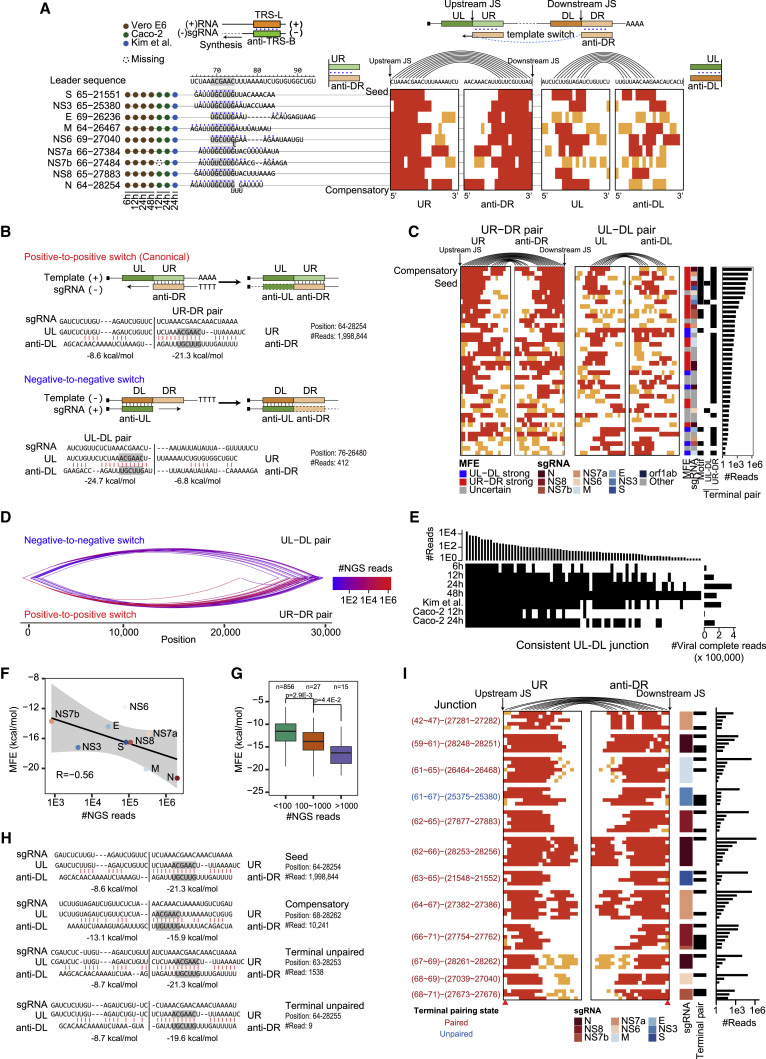
Coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is currently a global pandemic. CoVs are known to generate negative subgenomes (subgenomic RNAs [sgRNAs]) through transcription-regulating sequence (TRS)-dependent template switching, but the global dynamic landscapes of coronaviral subgenomes and regulatory rules remain unclear. Here, using next-generation sequencing (NGS) short-read and Nanopore long-read poly(A) RNA sequencing in two cell types at multiple time points after infection with SARS-CoV-2, we identified hundreds of template switches and constructed the dynamic landscapes of SARS-CoV-2 subgenomes. Interestingly, template switching could occur in a bidirectional manner, with diverse SARS-CoV-2 subgenomes generated from successive template-switching events. The majority of template switches result from RNA-RNA interactions, including seed and compensatory modes, with terminal pairing status as a key determinant. Two TRS-independent template switch modes are also responsible for subgenome biogenesis. Our findings reveal the subgenome landscape of SARS-CoV-2 and its regulatory features, providing a molecular basis for understanding subgenome biogenesis and developing novel anti-viral strategies.
Keywords: COVID-19; Nanopore sequencing; RNA pairing; SARS-CoV-2; biogenesis; coronavirus; sgRNA; subgenome; template switch.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
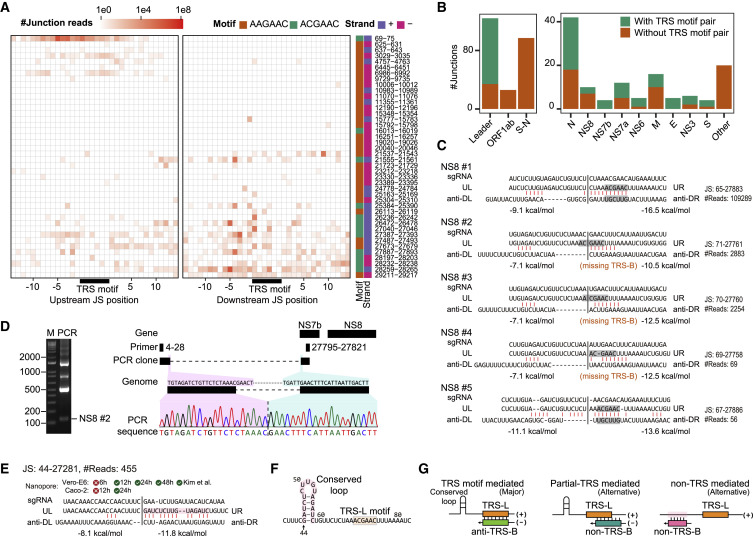
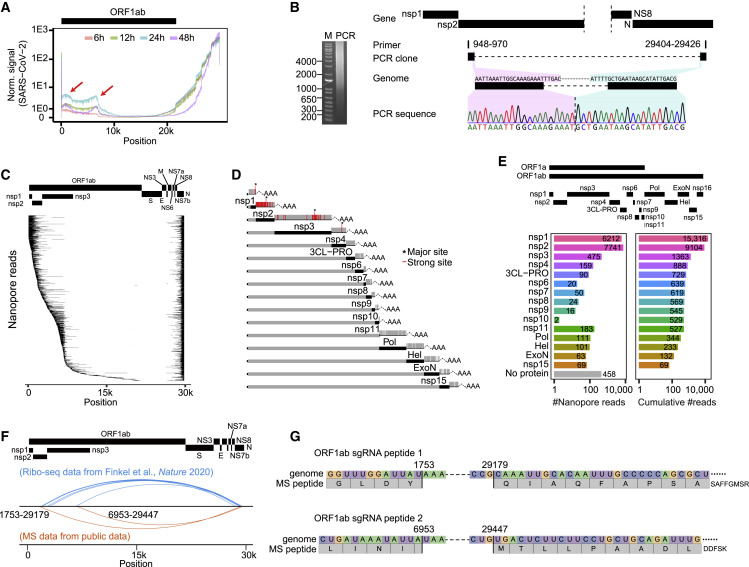
Similar articles
-
Pervasive generation of non-canonical subgenomic RNAs by SARS-CoV-2.Genome Med. 2020 Dec 1;12(1):108. doi: 10.1186/s13073-020-00802-w. Genome Med. 2020. PMID: 33256807 Free PMC article.
-
Transcriptional and epi-transcriptional dynamics of SARS-CoV-2 during cellular infection.Cell Rep. 2021 May 11;35(6):109108. doi: 10.1016/j.celrep.2021.109108. Epub 2021 Apr 23. Cell Rep. 2021. PMID: 33961822 Free PMC article.
-
Host-Virus Chimeric Events in SARS-CoV-2-Infected Cells Are Infrequent and Artifactual.J Virol. 2021 Jul 12;95(15):e0029421. doi: 10.1128/JVI.00294-21. Epub 2021 Jul 12. J Virol. 2021. PMID: 33980601 Free PMC article.
-
SARS-CoV-2 Subgenomic RNAs: Characterization, Utility, and Perspectives.Viruses. 2021 Sep 24;13(10):1923. doi: 10.3390/v13101923. Viruses. 2021. PMID: 34696353 Free PMC article. Review.
-
SARS-CoV-2 Sequencing for Variant Surveillance.Methods Mol Biol. 2023;2621:279-292. doi: 10.1007/978-1-0716-2950-5_16. Methods Mol Biol. 2023. PMID: 37041451 Review.
Cited by
-
Potential Role of the Antidepressants Fluoxetine and Fluvoxamine in the Treatment of COVID-19.Int J Mol Sci. 2022 Mar 30;23(7):3812. doi: 10.3390/ijms23073812. Int J Mol Sci. 2022. PMID: 35409171 Free PMC article. Review.
-
Nanopore sequencing technology and its applications.MedComm (2020). 2023 Jul 10;4(4):e316. doi: 10.1002/mco2.316. eCollection 2023 Aug. MedComm (2020). 2023. PMID: 37441463 Free PMC article. Review.
-
RNA architecture of porcine deltacoronavirus genome inside virions detected by vRIC-seq.Sci Data. 2024 Oct 14;11(1):1124. doi: 10.1038/s41597-024-03975-w. Sci Data. 2024. PMID: 39402053 Free PMC article.
-
Translation landscape of SARS-CoV-2 noncanonical subgenomic RNAs.Virol Sin. 2022 Dec;37(6):813-822. doi: 10.1016/j.virs.2022.09.003. Epub 2022 Sep 6. Virol Sin. 2022. PMID: 36075564 Free PMC article.
-
An Overview of Fluvoxamine and its Use in SARS-CoV-2 Treatment.Cureus. 2023 Jan 24;15(1):e34158. doi: 10.7759/cureus.34158. eCollection 2023 Jan. Cureus. 2023. PMID: 36843775 Free PMC article. Review.
References
-
- Davidson A.D., Williamson M.K., Lewis S., Shoemark D., Carroll M.W., Heesom K.J., Zambon M., Ellis J., Lewis P.A., Hiscox J.A., Matthews D.A. Characterisation of the transcriptome and proteome of SARS-CoV-2 reveals a cell passage induced in-frame deletion of the furin-like cleavage site from the spike glycoprotein. Genome Med. 2020;12:68. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
