

Upstream Regulators of Fibroblast Growth Factor 23
- PMID: 33716961
- PMCID: PMC7952762
- DOI: 10.3389/fendo.2021.588096
Upstream Regulators of Fibroblast Growth Factor 23
Abstract
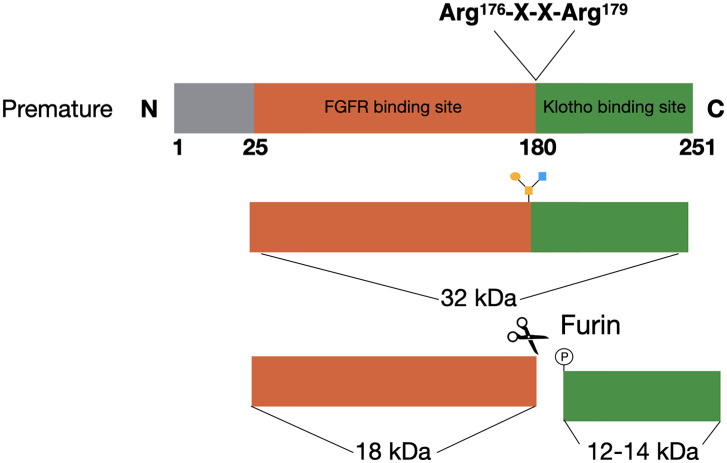
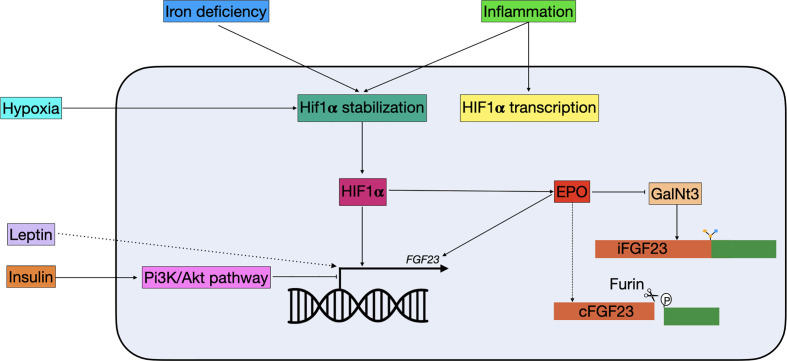
Fibroblast growth factor 23 (FGF23) has been described as an important regulator of mineral homeostasis, but has lately also been linked to iron deficiency, inflammation, and erythropoiesis. FGF23 is essential for the maintenance of phosphate homeostasis in the body and activating mutations in the gene itself or inactivating mutations in its upstream regulators can result in severe chronic hypophosphatemia, where an unbalanced mineral homeostasis often leads to rickets in children and osteomalacia in adults. FGF23 can be regulated by changes in transcriptional activity or by changes at the post-translational level. The balance between O-glycosylation and phosphorylation is an important determinant of how much active intact or inactive cleaved FGF23 will be released in the circulation. In the past years, it has become evident that iron deficiency and inflammation regulate FGF23 in a way that is not associated with its classical role in mineral metabolism. These conditions will not only result in an upregulation of FGF23 transcription, but also in increased cleavage, leaving the levels of active intact FGF23 unchanged. The exact mechanisms behind and function of this process are still unclear. However, a deeper understanding of FGF23 regulation in both the classical and non-classical way is important to develop better treatment options for diseases associated with disturbed FGF23 biology. In this review, we describe how the currently known upstream regulators of FGF23 change FGF23 transcription and affect its post-translational modifications at the molecular level.
Keywords: FGF23; erythropoiesis; hypoxia; inflammation; iron deficiency; osteocytes; phosphate; vitamin D.
Copyright © 2021 Ratsma, Zillikens and van der Eerden.
Conflict of interest statement
The authors declare that the research was conducted without any commercial or financial relationships that may be regarded as a potential conflict of interest.
Figures

Similar articles
-
FGF23 and Hypophosphatemic Rickets/Osteomalacia.Curr Osteoporos Rep. 2021 Dec;19(6):669-675. doi: 10.1007/s11914-021-00709-4. Epub 2021 Nov 10. Curr Osteoporos Rep. 2021. PMID: 34755323 Review.
-
Pathogenic role of Fgf23 in Dmp1-null mice.Am J Physiol Endocrinol Metab. 2008 Aug;295(2):E254-61. doi: 10.1152/ajpendo.90201.2008. Epub 2008 Jun 17. Am J Physiol Endocrinol Metab. 2008. PMID: 18559986 Free PMC article.
-
FGF23 at the crossroads of phosphate, iron economy and erythropoiesis.Nat Rev Nephrol. 2020 Jan;16(1):7-19. doi: 10.1038/s41581-019-0189-5. Epub 2019 Sep 13. Nat Rev Nephrol. 2020. PMID: 31519999 Review.
-
Advances in understanding of phosphate homeostasis and related disorders.Endocr J. 2022 Aug 29;69(8):881-896. doi: 10.1507/endocrj.EJ22-0239. Epub 2022 Jul 13. Endocr J. 2022. PMID: 35831119 Review.
-
X-Linked Hypophosphatemia and FGF23-Related Hypophosphatemic Diseases: Prospect for New Treatment.Endocr Rev. 2018 Jun 1;39(3):274-291. doi: 10.1210/er.2017-00220. Endocr Rev. 2018. PMID: 29381780 Review.
Cited by
-
New developments in the biology of fibroblast growth factors.WIREs Mech Dis. 2022 Jul;14(4):e1549. doi: 10.1002/wsbm.1549. Epub 2022 Feb 9. WIREs Mech Dis. 2022. PMID: 35142107 Free PMC article. Review.
-
The role of fibroblast growth factor 23 in regulation of phosphate balance.Pediatr Nephrol. 2024 Dec;39(12):3439-3451. doi: 10.1007/s00467-024-06395-5. Epub 2024 Jun 14. Pediatr Nephrol. 2024. PMID: 38874635 Review.
-
Tumor-Induced Osteomalacia in a Patient with Crohn's Disease: A Case Report and Approach to Investigating Hypophosphatemia.Case Rep Gastroenterol. 2024 Feb 26;18(1):81-89. doi: 10.1159/000536136. eCollection 2024 Jan-Dec. Case Rep Gastroenterol. 2024. PMID: 38410687 Free PMC article.
-
Iron and Bone Pathophysiology.Adv Exp Med Biol. 2025;1480:311-325. doi: 10.1007/978-3-031-92033-2_20. Adv Exp Med Biol. 2025. PMID: 40603799 Review.
-
In-center Nocturnal Hemodialysis Reduced the Circulating FGF23, Left Ventricular Hypertrophy, and All-Cause Mortality: A Retrospective Cohort Study.Front Med (Lausanne). 2022 Jun 21;9:912764. doi: 10.3389/fmed.2022.912764. eCollection 2022. Front Med (Lausanne). 2022. PMID: 35801203 Free PMC article.
References
-
- Prader A, Illig R, Uehlinger E, Stalder G. Rickets following bone tumor. Helv Paediatr Acta (1959) 14:554–65. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous