

Equivolumetric Protocol Generates Library Sizes Proportional to Total Microbial Load in 16S Amplicon Sequencing
- PMID: 33717032
- PMCID: PMC7952455
- DOI: 10.3389/fmicb.2021.638231
Equivolumetric Protocol Generates Library Sizes Proportional to Total Microbial Load in 16S Amplicon Sequencing
Abstract
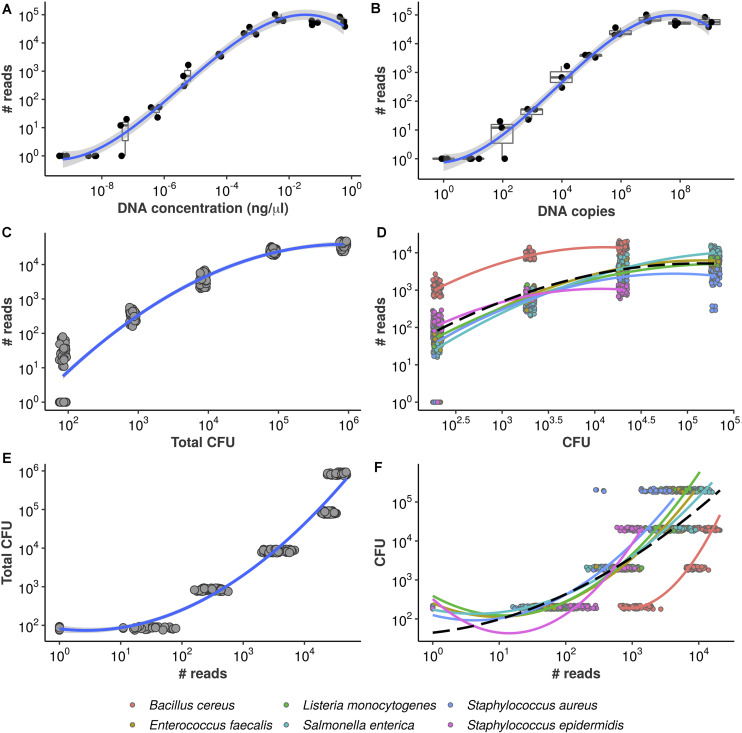
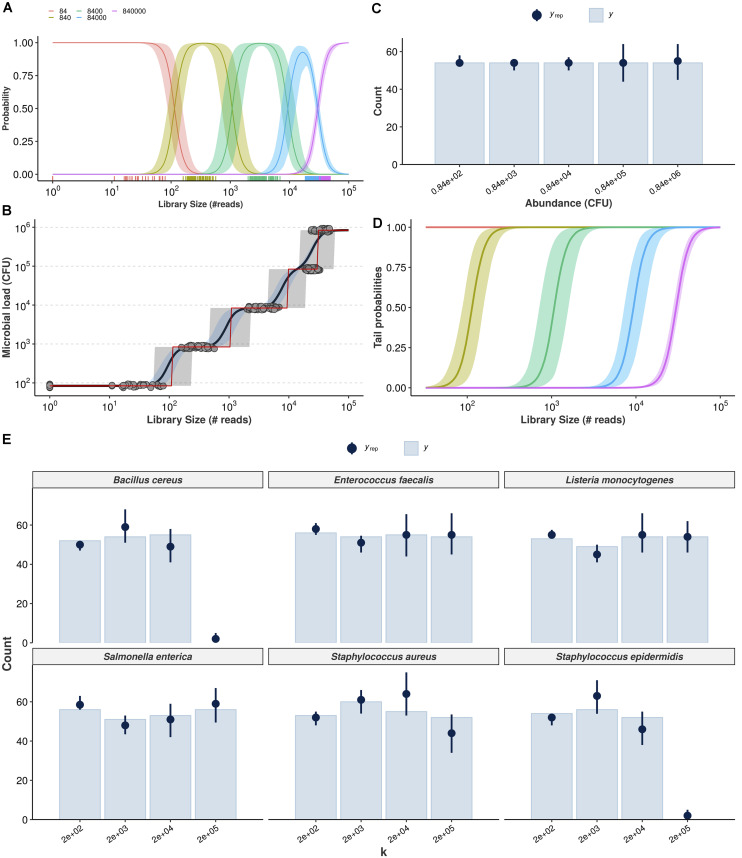
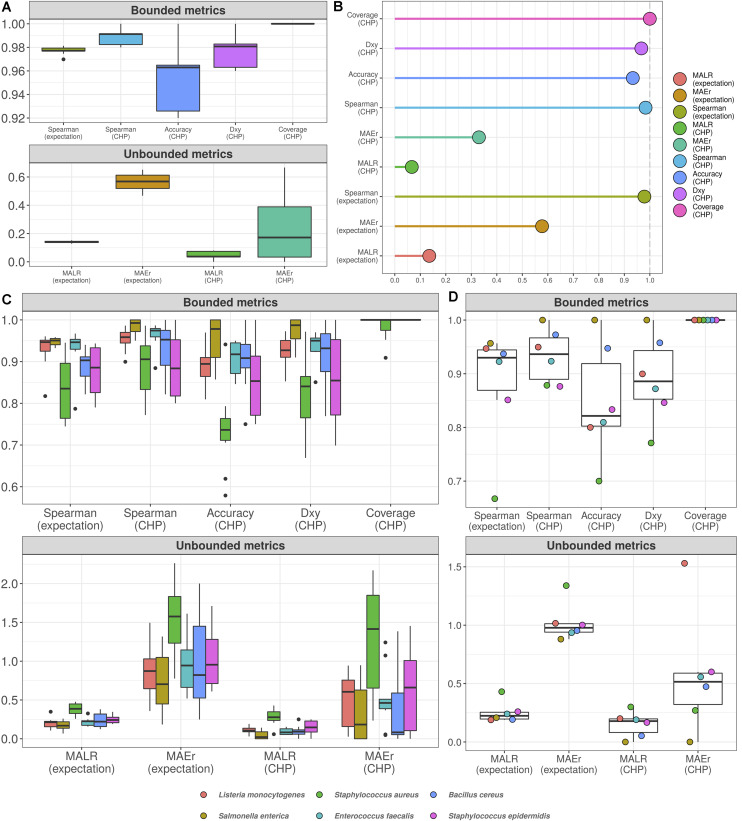
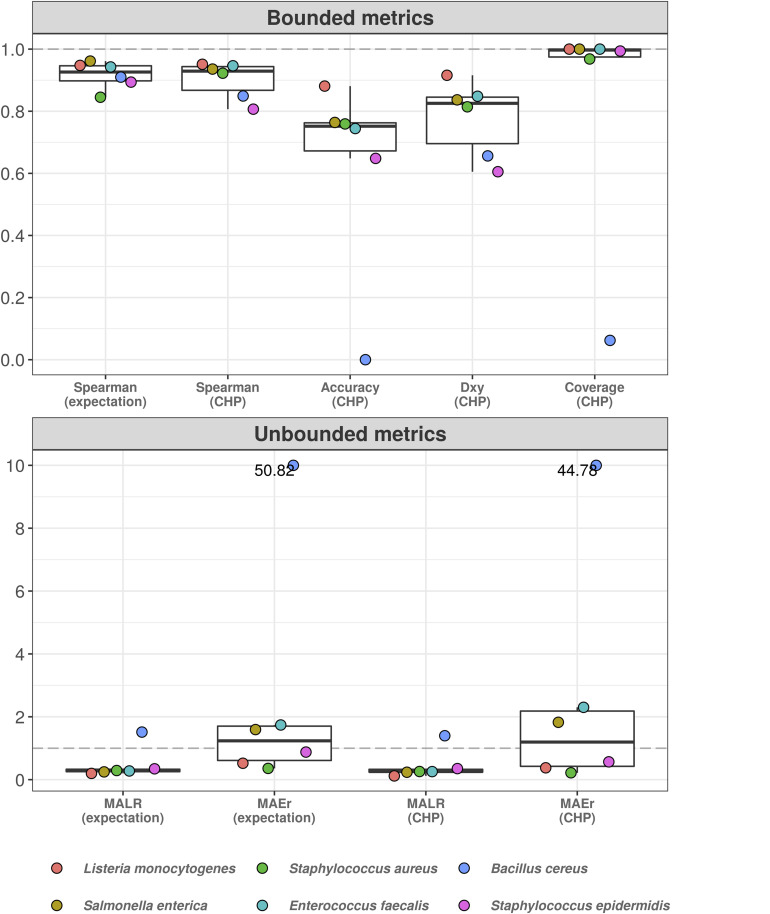
High-throughput sequencing of 16S rRNA amplicon has been extensively employed to perform microbiome characterization worldwide. As a culture-independent methodology, it has allowed high-level profiling of sample bacterial composition directly from samples. However, most studies are limited to information regarding relative bacterial abundances (sample proportions), ignoring scenarios in which sample microbe biomass can vary widely. Here, we use an equivolumetric protocol for 16S rRNA amplicon library preparation capable of generating Illumina sequencing data responsive to input DNA, recovering proportionality between observed read counts and absolute bacterial abundances within each sample. Under specified conditions, we show that the estimation of colony-forming units (CFU), the most common unit of bacterial abundance in classical microbiology, is challenged mostly by resolution and taxon-to-taxon variation. We propose Bayesian cumulative probability models to address such issues. Our results indicate that predictive errors vary consistently below one order of magnitude for total microbial load and abundance of observed bacteria. We also demonstrate our approach has the potential to generalize to previously unseen bacteria, but predictive performance is hampered by specific taxa of uncommon profile. Finally, it remains clear that high-throughput sequencing data are not inherently restricted to sample proportions only, and such technologies bear the potential to meet the working scales of traditional microbiology.
Keywords: 16S rRNA; Illumina; absolute abundances; amplicon sequencing; bacteria; colony-forming units; microbiome.
Copyright © 2021 Cruz, Christoff and de Oliveira.
Conflict of interest statement
All authors are currently full-time employees of BiomeHub (SC, Brazil), a research and consulting company specialized in microbiome technologies. BiomeHub funded the study design, analysis, and data submission for publication.
Figures

References
-
- Agresti A. (2010a). Wiley Series in Probability and Statistics. Hoboken, NJ: John Wiley & Sons, Inc, 281–314. 10.1002/9780470594001.ch10 - DOI
-
- Agresti A. (2010b). Wiley Series in Probability and Statistics. Hoboken, NJ: John Wiley & Sons, Inc, 44–87. 10.1002/9780470594001.ch3 - DOI
-
- Bürkner P. (2017). brms?: an R Package for bayesian multilevel models using stan. J. Stat. Softw. 80:29856. 10.18637/jss.v080.i01 - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials