

Immunological Feature and Transcriptional Signaling of Ly6C Monocyte Subsets From Transcriptome Analysis in Control and Hyperhomocysteinemic Mice
- PMID: 33717169
- PMCID: PMC7947624
- DOI: 10.3389/fimmu.2021.632333
Immunological Feature and Transcriptional Signaling of Ly6C Monocyte Subsets From Transcriptome Analysis in Control and Hyperhomocysteinemic Mice
Abstract
Background: Murine monocytes (MC) are classified into Ly6Chigh and Ly6Clow MC. Ly6Chigh MC is the pro-inflammatory subset and the counterpart of human CD14++CD16+ intermediate MC which contributes to systemic and tissue inflammation in various metabolic disorders, including hyperhomocysteinemia (HHcy). This study aims to explore molecule signaling mediating MC subset differentiation in HHcy and control mice.
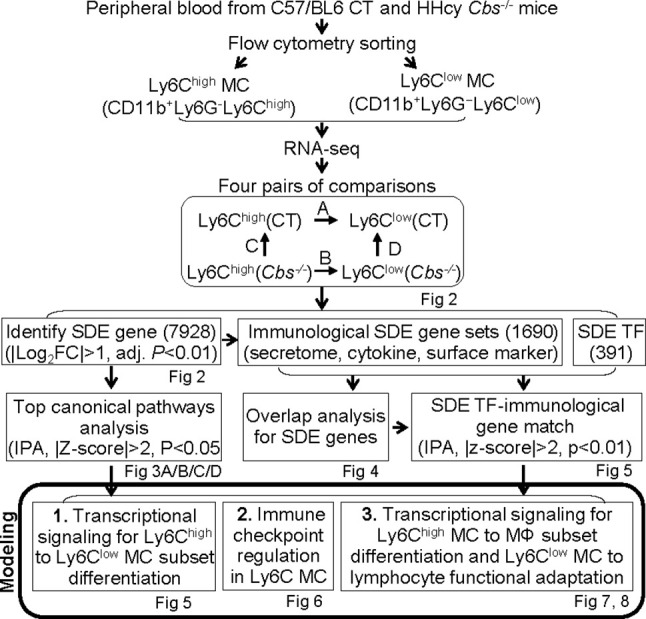
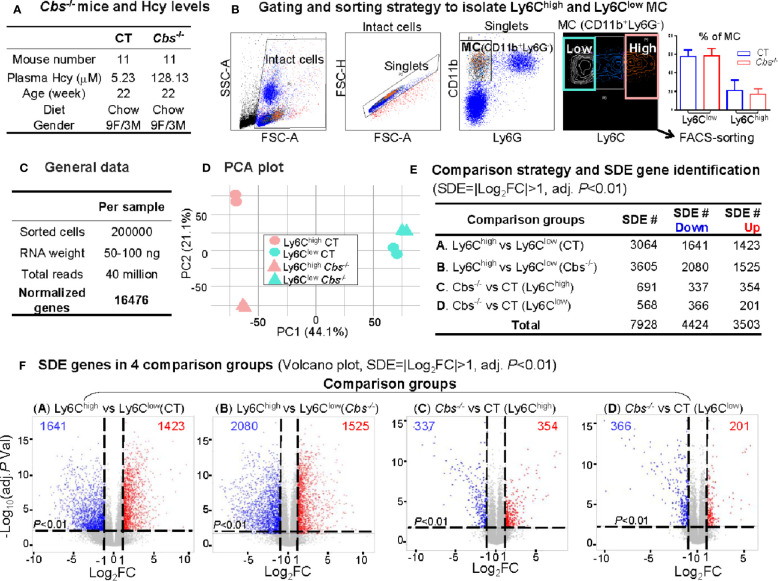
Methods: RNA-seq was performed in blood Ly6Chigh and Ly6Clow MC sorted by flow cytometry from control and HHcy cystathionine β-synthase gene-deficient (Cbs-/-) mice. Transcriptome data were analyzed by comparing Ly6Chigh vs. Ly6Clow in control mice, Ly6Chigh vs. Ly6Clow in Cbs-/- mice, Cbs-/- Ly6Chigh vs. control Ly6Chigh MC and Cbs-/- Ly6Clow vs. control Ly6Clow MC by using intensive bioinformatic strategies. Significantly differentially expressed (SDE) immunological genes and transcription factor (TF) were selected for functional pathways and transcriptional signaling identification.
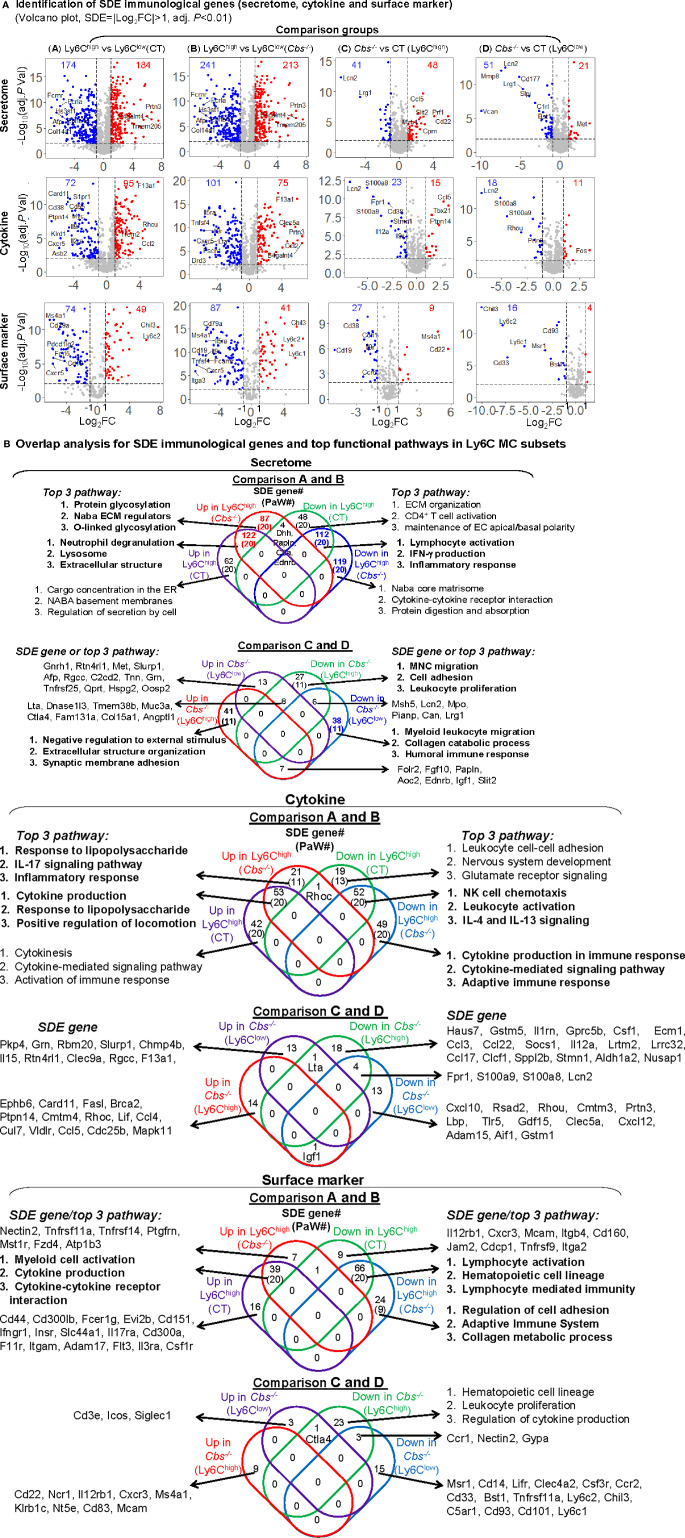
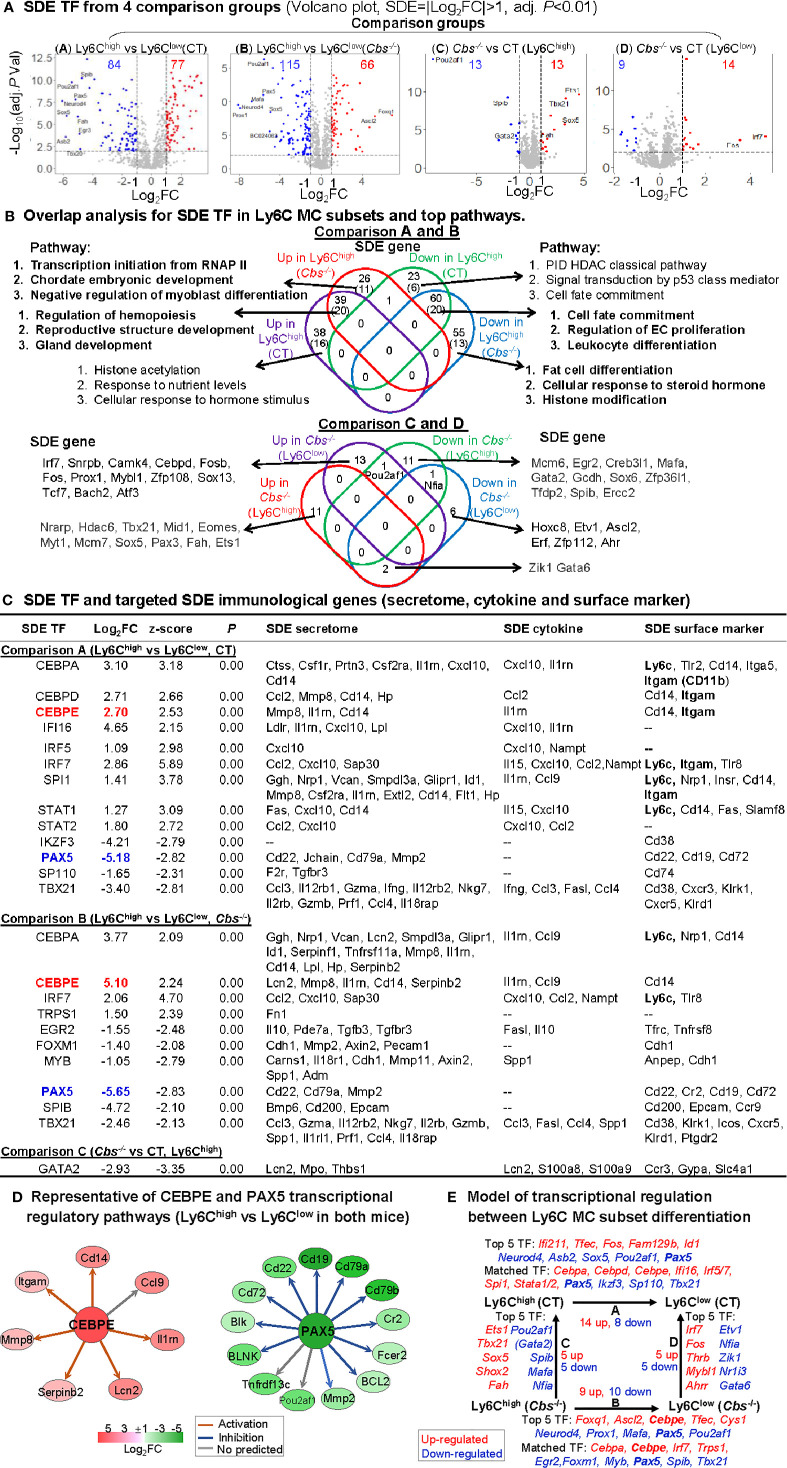
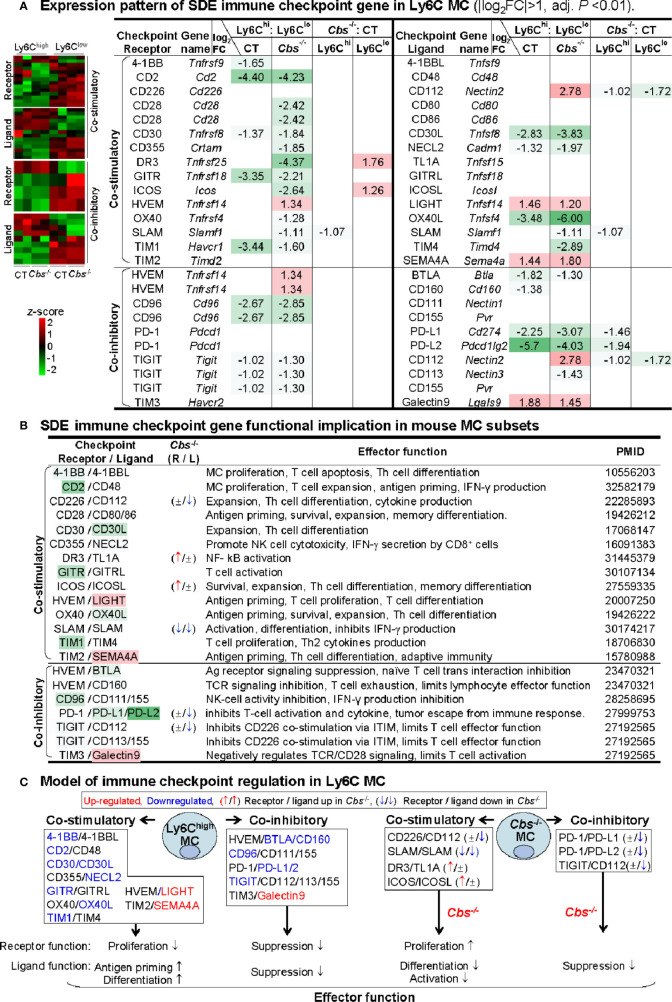
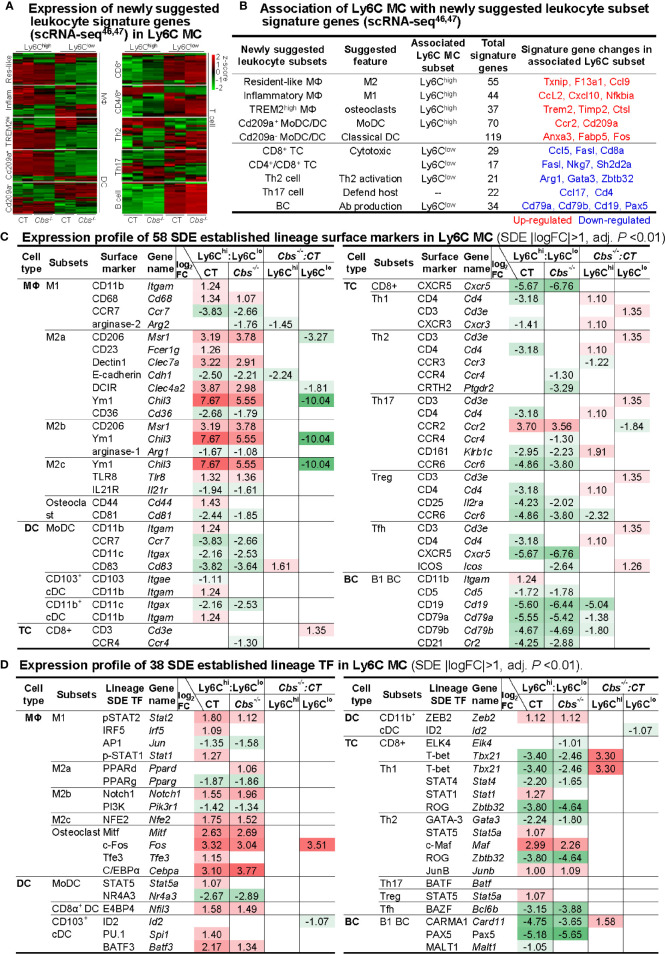
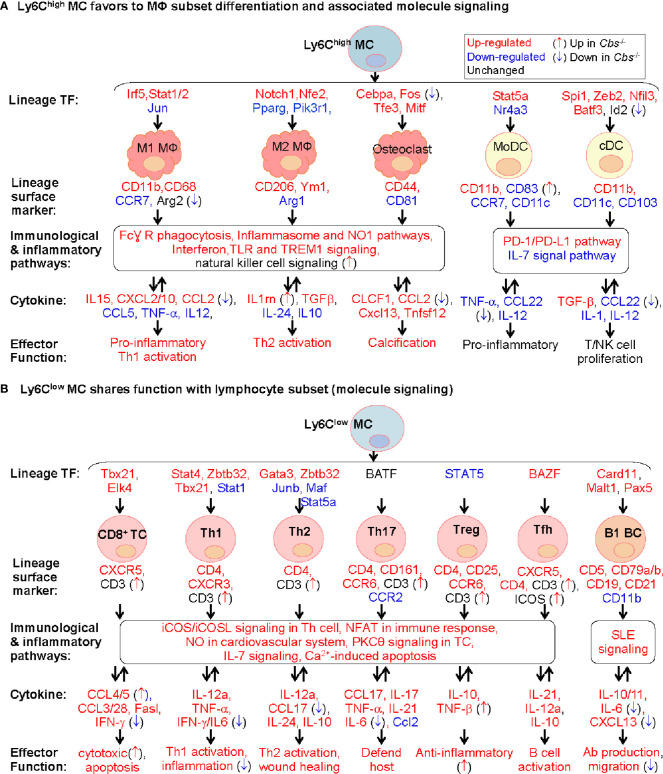
Results: A total of 7,928 SDE genes and 46 canonical pathways derived from it were identified. Ly6Chigh MC exhibited activated neutrophil degranulation, lysosome, cytokine production/receptor interaction and myeloid cell activation pathways, and Ly6Clow MC presented features of lymphocyte immunity pathways in both mice. Twenty-four potential transcriptional regulatory pathways were identified based on SDE TFs matched with their corresponding SDE immunological genes. Ly6Chigh MC presented downregulated co-stimulatory receptors (CD2, GITR, and TIM1) which direct immune cell proliferation, and upregulated co-stimulatory ligands (LIGHT and SEMA4A) which trigger antigen priming and differentiation. Ly6Chigh MC expressed higher levels of macrophage (MΦ) markers, whereas, Ly6Clow MC highly expressed lymphocyte markers in both mice. HHcy in Cbs-/- mice reinforced inflammatory features in Ly6Chigh MC by upregulating inflammatory TFs (Ets1 and Tbx21) and strengthened lymphocytes functional adaptation in Ly6Clow MC by increased expression of CD3, DR3, ICOS, and Fos. Finally, we established 3 groups of transcriptional models to describe Ly6Chigh to Ly6Clow MC subset differentiation, immune checkpoint regulation, Ly6Chigh MC to MΦ subset differentiation and Ly6Clow MC to lymphocyte functional adaptation.
Conclusions: Ly6Chigh MC displayed enriched inflammatory pathways and favored to be differentiated into MΦ. Ly6Clow MC manifested activated T-cell signaling pathways and potentially can adapt the function of lymphocytes. HHcy reinforced inflammatory feature in Ly6Chigh MC and strengthened lymphocytes functional adaptation in Ly6Clow MC.
Keywords: hyperhomocysteinemia; immune checkpoint; immunological gene; locus C (Ly6C) monocyte subset; lymphocyte antigen 6 complex; transcription factor.
Copyright © 2021 Yang, Liu, Sun, Fang, Snyder, Saredy, Ji, Shen, Qin, Wu, Yang and Wang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

Similar articles
-
Adaptive Immune Response Signaling Is Suppressed in Ly6Chigh Monocyte but Upregulated in Monocyte Subsets of ApoE-/- Mice - Functional Implication in Atherosclerosis.Front Immunol. 2021 Dec 20;12:809208. doi: 10.3389/fimmu.2021.809208. eCollection 2021. Front Immunol. 2021. PMID: 34987524 Free PMC article.
-
Severe hyperhomocysteinemia promotes bone marrow-derived and resident inflammatory monocyte differentiation and atherosclerosis in LDLr/CBS-deficient mice.Circ Res. 2012 Jun 22;111(1):37-49. doi: 10.1161/CIRCRESAHA.112.269472. Epub 2012 May 24. Circ Res. 2012. PMID: 22628578 Free PMC article.
-
Hyperhomocysteinemia promotes inflammatory monocyte generation and accelerates atherosclerosis in transgenic cystathionine beta-synthase-deficient mice.Circulation. 2009 Nov 10;120(19):1893-902. doi: 10.1161/CIRCULATIONAHA.109.866889. Epub 2009 Oct 26. Circulation. 2009. PMID: 19858416 Free PMC article.
-
Metabolism-associated danger signal-induced immune response and reverse immune checkpoint-activated CD40+ monocyte differentiation.J Hematol Oncol. 2017 Jul 24;10(1):141. doi: 10.1186/s13045-017-0504-1. J Hematol Oncol. 2017. PMID: 28738836 Free PMC article. Review.
-
Harnessing monocyte dynamics for treatment of multiple sclerosis; insights from experimental model studies.Immunother Adv. 2025 Apr 20;5(1):ltaf003. doi: 10.1093/immadv/ltaf003. eCollection 2025. Immunother Adv. 2025. PMID: 40342728 Free PMC article. Review.
Cited by
-
Identification of Genes Associated with Decreasing Abundance of Monocytes in Long-Term Peritoneal Dialysis Patients.Int J Gen Med. 2023 Nov 3;16:5017-5030. doi: 10.2147/IJGM.S435041. eCollection 2023. Int J Gen Med. 2023. PMID: 37942472 Free PMC article.
-
Classical monocyte-derived macrophages as therapeutic targets of umbilical cord mesenchymal stem cells: comparison of intratracheal and intravenous administration in a mouse model of pulmonary fibrosis.Respir Res. 2023 Mar 5;24(1):68. doi: 10.1186/s12931-023-02357-x. Respir Res. 2023. PMID: 36870972 Free PMC article.
-
NSC243928 Treatment Induces Anti-Tumor Immune Response in Mouse Mammary Tumor Models.Cancers (Basel). 2023 Feb 25;15(5):1468. doi: 10.3390/cancers15051468. Cancers (Basel). 2023. PMID: 36900259 Free PMC article.
-
Small Heterodimer Partner Modulates Macrophage Differentiation during Innate Immune Response through the Regulation of Peroxisome Proliferator Activated Receptor Gamma, Mitogen-Activated Protein Kinase, and Nuclear Factor Kappa B Pathways.Biomedicines. 2023 Aug 28;11(9):2403. doi: 10.3390/biomedicines11092403. Biomedicines. 2023. PMID: 37760844 Free PMC article.
-
Macrophages producing chondroitin sulfate proteoglycan-4 induce neuro-cardiac junction impairment in Duchenne muscular dystrophy.J Pathol. 2025 Jan;265(1):1-13. doi: 10.1002/path.6362. Epub 2024 Nov 11. J Pathol. 2025. PMID: 39523812 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous