

Mitochondrial Damage-Induced Innate Immune Activation in Vascular Smooth Muscle Cells Promotes Chronic Kidney Disease-Associated Plaque Vulnerability
- PMID: 33717842
- PMCID: PMC7927614
- DOI: 10.1002/advs.202002738
Mitochondrial Damage-Induced Innate Immune Activation in Vascular Smooth Muscle Cells Promotes Chronic Kidney Disease-Associated Plaque Vulnerability
Abstract
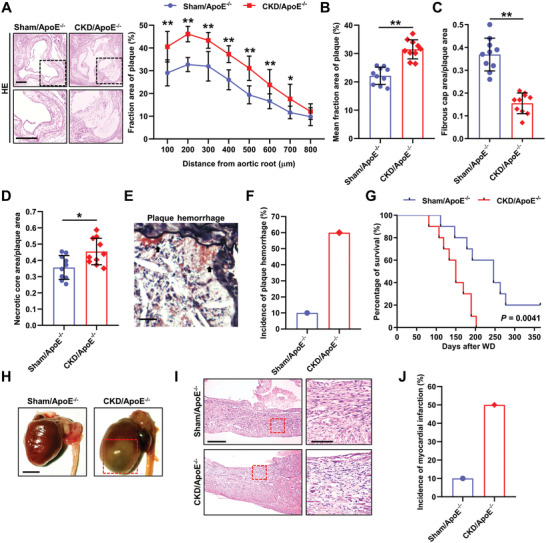
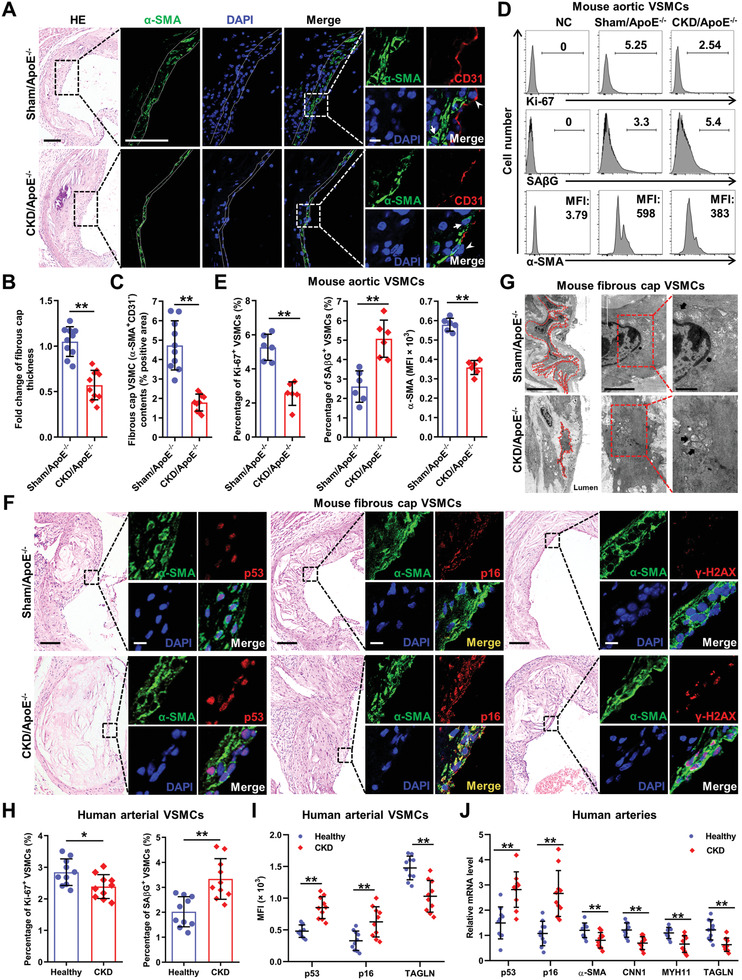
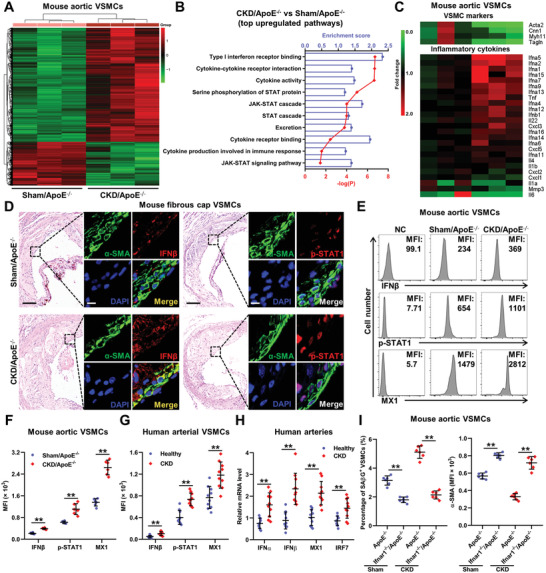
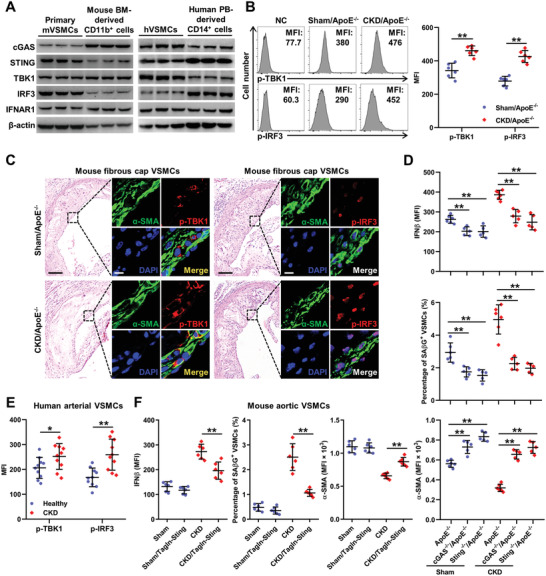
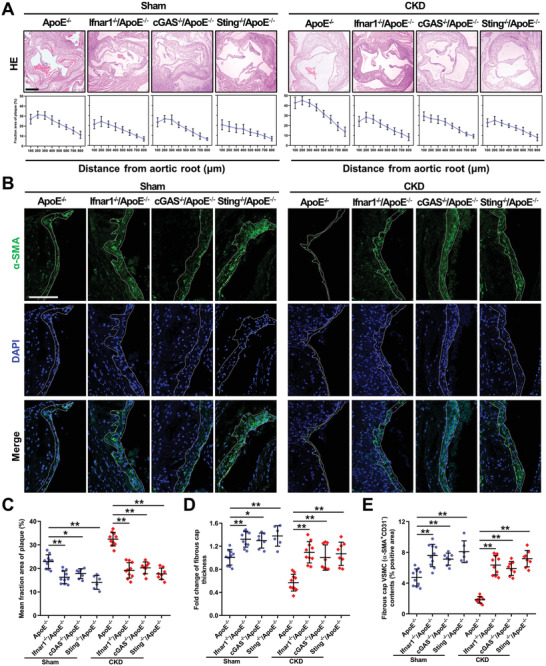
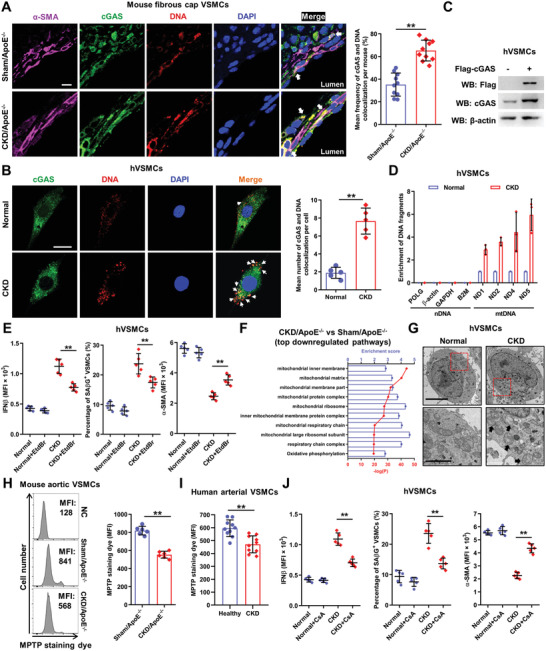
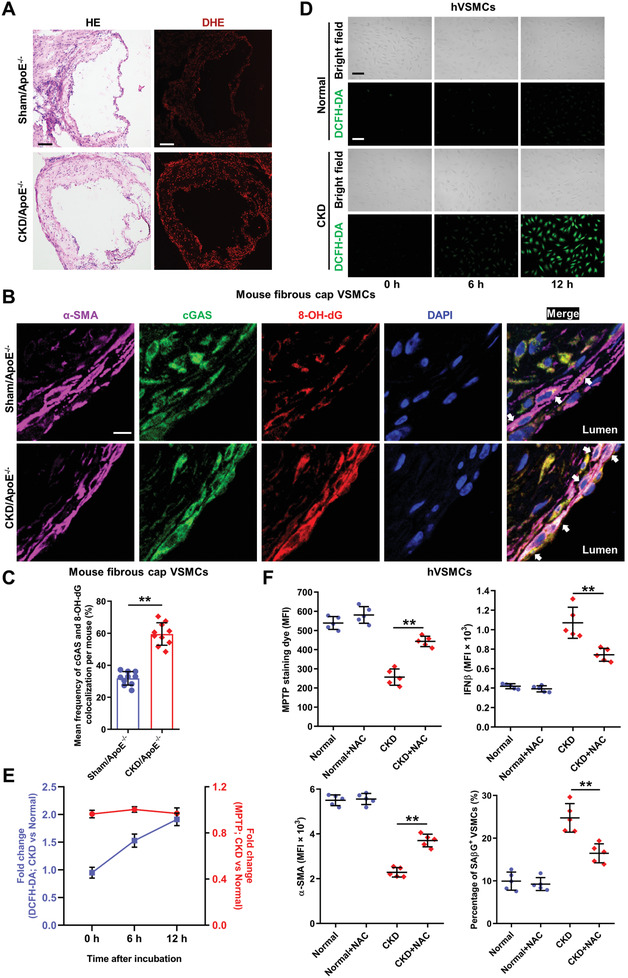
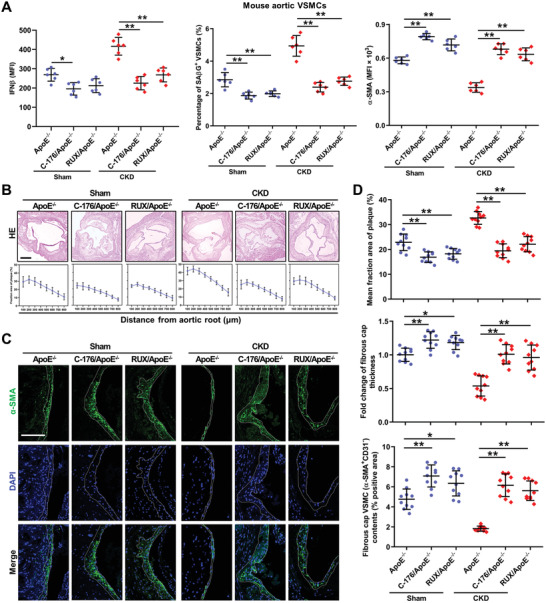
Chronic kidney disease (CKD) is associated with accelerated atherosclerosis progression and high incidence of cardiovascular events, hinting that atherosclerotic plaques in CKD may be vulnerable. However, its cause and mechanism remain obscure. Here, it is shown that apolipoprotein E-deficient (ApoE-/-) mouse with CKD (CKD/ApoE-/- mouse) is a useful model for investigating the pathogenesis of plaque vulnerability, and premature senescence and phenotypic switching of vascular smooth muscle cells (VSMCs) contributes to CKD-associated plaque vulnerability. Subsequently, VSMC phenotypes in patients with CKD and CKD/ApoE-/- mice are comprehensively investigated. Using multi-omics analysis and targeted and VSMC-specific gene knockout mice, VSMCs are identified as both type-I-interferon (IFN-I)-responsive and IFN-I-productive cells. Mechanistically, mitochondrial damage resulting from CKD-induced oxidative stress primes the cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathway to trigger IFN-I response in VSMCs. Enhanced IFN-I response then induces VSMC premature senescence and phenotypic switching in an autocrine/paracrine manner, resulting in the loss of fibrous cap VSMCs and fibrous cap thinning. Conversely, blocking IFN-I response remarkably attenuates CKD-associated plaque vulnerability. These findings reveal that IFN-I response in VSMCs through immune sensing of mitochondrial damage is essential for the pathogenesis of CKD-associated plaque vulnerability. Mitigating IFN-I response may hold promise for the treatment of CKD-associated cardiovascular diseases.
Keywords: atherosclerosis; chronic kidney disease; cyclic GMP‐AMP synthase‐stimulator of interferon genes pathway; plaque vulnerability; vascular smooth muscle cell.
© 2021 The Authors. Advanced Science published by Wiley‐VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Jha V., Garcia‐Garcia G., Iseki K., Li Z., Naicker S., Plattner B., Saran R., Wang A. Y.‐M., Yang C.‐W., Lancet 2013, 382, 260. - PubMed
-
- Drüeke T. B., Massy Z. A., Nat. Rev. Nephrol. 2010, 6, 723. - PubMed
-
- Bro S., J. Am. Soc. Nephrol. 2003, 14, 2466. - PubMed
-
- Yang K., Du C., Wang X., Li F., Xu Y., Wang S., Chen S., Chen F., Shen M., Chen M., Hu M., He T., Su Y., Wang J., Zhao J., Blood 2017, 129, 2667. - PubMed
-
- Libby P., Pasterkamp G., Eur. Heart J. 2015, 36, 2984. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous