

Phylogeography of Human and Animal Coxiella burnetii Strains: Genetic Fingerprinting of Q Fever in Belgium
- PMID: 33718257
- PMCID: PMC7952626
- DOI: 10.3389/fcimb.2020.625576
Phylogeography of Human and Animal Coxiella burnetii Strains: Genetic Fingerprinting of Q Fever in Belgium
Abstract
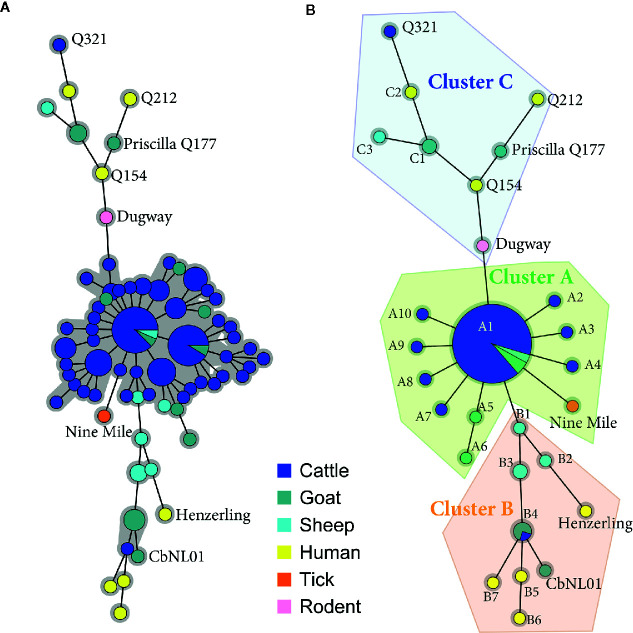
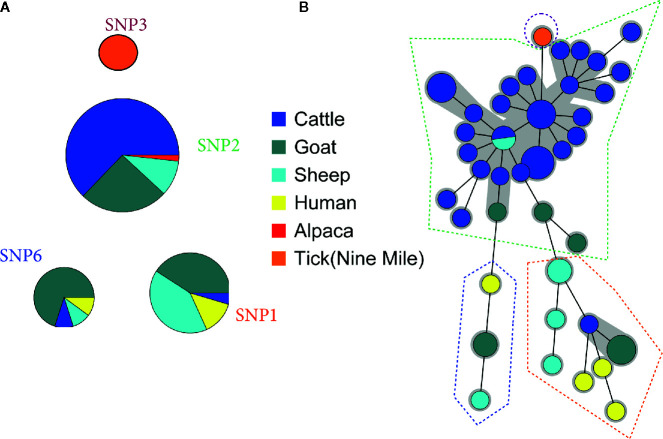
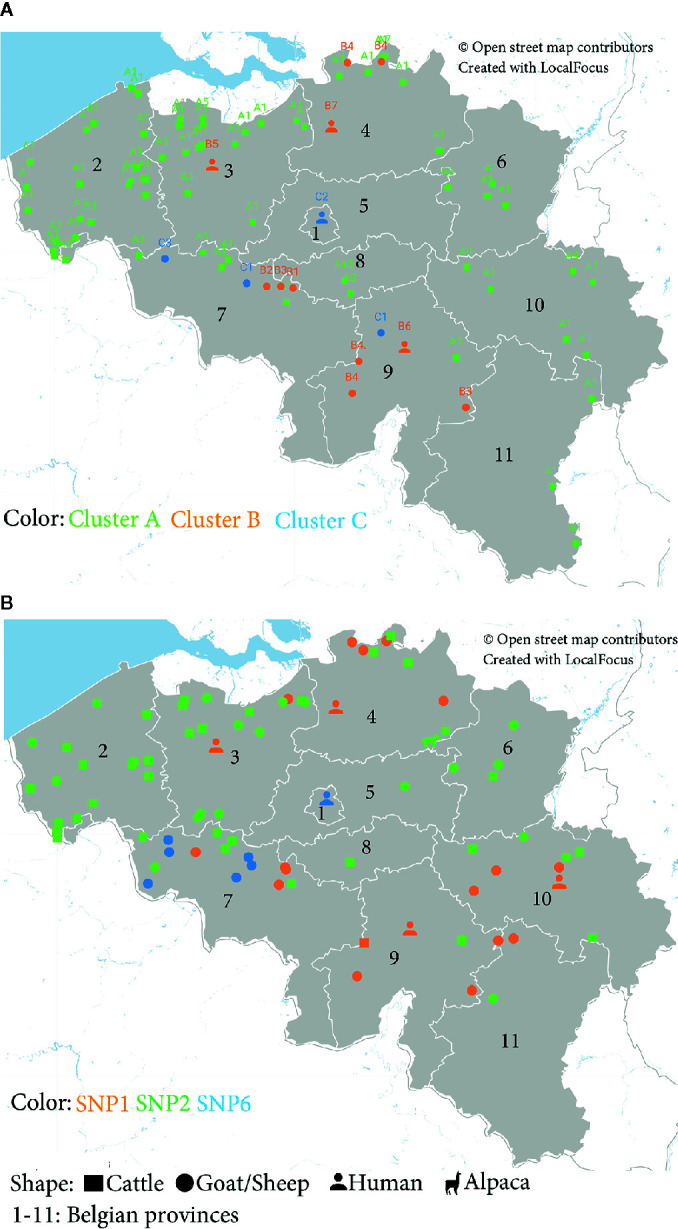
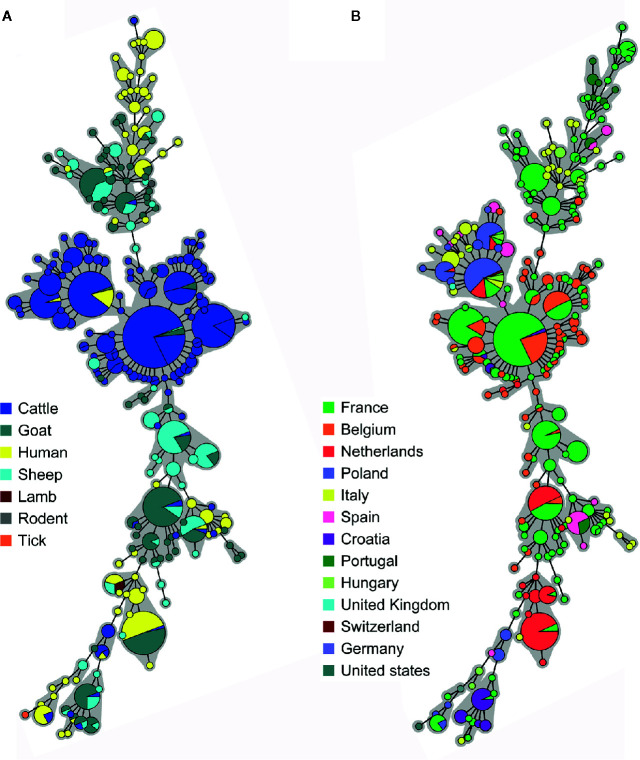
Q fever is a zoonotic disease caused by the bacteria Coxiella burnetii. Domestic ruminants are the primary source for human infection, and the identification of likely contamination routes from the reservoir animals the critical point to implement control programs. This study shows that Q fever is detected in Belgium in abortion of cattle, goat and sheep at a different degree of apparent prevalence (1.93%, 9.19%, and 5.50%, respectively). In addition, and for the first time, it is detected in abortion of alpaca (Vicugna pacos), raising questions on the role of these animals as reservoirs. To determine the relationship between animal and human strains, Multiple Locus Variable-number Tandem Repeat Analysis (MLVA) (n=146), Single-Nucleotide Polymorphism (SNP) (n=92) and Whole Genome Sequencing (WGS) (n=4) methods were used to characterize samples/strains during 2009-2019. Three MLVA clusters (A, B, C) subdivided in 23 subclusters (A1-A12, B1-B8, C1-C3) and 3 SNP types (SNP1, SNP2, SNP6) were identified. The SNP2 type/MLVA cluster A was the most abundant and dispersed genotype over the entire territory, but it seemed not responsible for human cases, as it was only present in animal samples. The SNP1/MLVA B and SNP6/MLVA C clusters were mostly found in small ruminant and human samples, with the rare possibility of spillovers in cattle. SNP1/MLVA B cluster was present in all Belgian areas, while the SNP6/MLVA C cluster appeared more concentrated in the Western provinces. A broad analysis of European MLVA profiles confirmed the host-species distribution described for Belgian samples. In silico genotyping (WGS) further identified the spacer types and the genomic groups of C. burnetii Belgian strains: cattle and goat SNP2/MLVA A isolates belonged to ST61 and genomic group III, while the goat SNP1/MLVA B strain was classified as ST33 and genomic group II. In conclusion, Q fever is widespread in all Belgian domestic ruminants and in alpaca. We determined that the public health risk in Belgium is likely linked to specific genomic groups (SNP1/MLVA B and SNP6/MLVA C) mostly found in small ruminant strains. Considering the concordance between Belgian and European results, these considerations could be extended to other European countries.
Keywords: Coxiella burnetii; MLVA; SNP; WGS; alpaca; animals; humans.
Copyright © 2021 Tomaiuolo, Boarbi, Fancello, Michel, Desqueper, Grégoire, Callens, Fretin, Devriendt, Cox and Mori.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Genotyping of Coxiella burnetii from domestic ruminants in northern Spain.BMC Vet Res. 2012 Dec 10;8:241. doi: 10.1186/1746-6148-8-241. BMC Vet Res. 2012. PMID: 23227921 Free PMC article.
-
MLVA and com1 genotyping of Coxiella burnetii in farmed ruminants in Great Britain.Vet Microbiol. 2023 Feb;277:109629. doi: 10.1016/j.vetmic.2022.109629. Epub 2022 Dec 9. Vet Microbiol. 2023. PMID: 36535174
-
Molecular epidemiology of Coxiella burnetii in French livestock reveals the existence of three main genotype clusters and suggests species-specific associations as well as regional stability.Infect Genet Evol. 2017 Mar;48:142-149. doi: 10.1016/j.meegid.2016.12.015. Epub 2016 Dec 20. Infect Genet Evol. 2017. PMID: 28007602
-
Q fever and seroprevalence of Coxiella burnetii in domestic ruminants.Vet Ital. 2018 Dec 31;54(4):265-279. doi: 10.12834/VetIt.1113.6046.3. Vet Ital. 2018. PMID: 30681125 Review.
-
Coxiella burnetii infections in sheep or goats: an opinionated review.Vet Microbiol. 2015 Dec 14;181(1-2):119-29. doi: 10.1016/j.vetmic.2015.07.011. Epub 2015 Jul 15. Vet Microbiol. 2015. PMID: 26315774 Review.
Cited by
-
A zoonotic cause of blood culture-negative infective endocarditis in Belgium: Case report and review of the literature on Q fever.IDCases. 2022 Aug 6;29:e01595. doi: 10.1016/j.idcr.2022.e01595. eCollection 2022. IDCases. 2022. PMID: 36032176 Free PMC article.
-
Genome-wide epitope mapping across multiple host species reveals significant diversity in antibody responses to Coxiella burnetii vaccination and infection.Front Immunol. 2023 Oct 26;14:1257722. doi: 10.3389/fimmu.2023.1257722. eCollection 2023. Front Immunol. 2023. PMID: 37954609 Free PMC article.
-
Metabolism and physiology of pathogenic bacterial obligate intracellular parasites.Front Cell Infect Microbiol. 2024 Mar 22;14:1284701. doi: 10.3389/fcimb.2024.1284701. eCollection 2024. Front Cell Infect Microbiol. 2024. PMID: 38585652 Free PMC article. Review.
-
Phenotype of Coxiella burnetii Strains of Different Sources and Genotypes in Bovine Mammary Gland Epithelial Cells.Pathogens. 2022 Nov 26;11(12):1422. doi: 10.3390/pathogens11121422. Pathogens. 2022. PMID: 36558755 Free PMC article.
-
Molecular detection of Coxiella burnetii in small ruminants and genotyping of specimens collected from goats in Poland.BMC Vet Res. 2021 Oct 28;17(1):341. doi: 10.1186/s12917-021-03051-0. BMC Vet Res. 2021. PMID: 34711239 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous