

A Simplified Amino Acidic Alphabet to Unveil the T-Cells Receptors Antigens: A Computational Perspective
- PMID: 33718327
- PMCID: PMC7947793
- DOI: 10.3389/fchem.2021.598802
A Simplified Amino Acidic Alphabet to Unveil the T-Cells Receptors Antigens: A Computational Perspective
Abstract
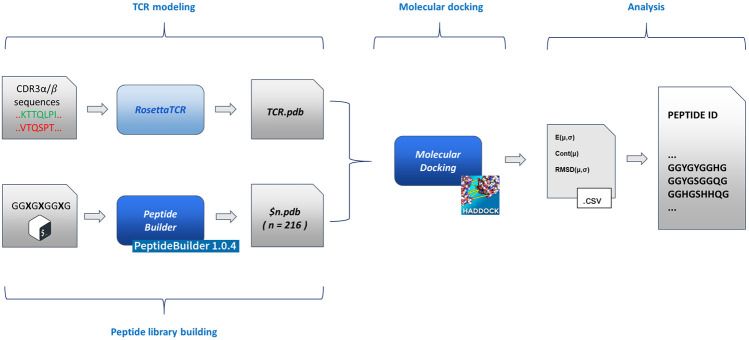
The exposure to pathogens triggers the activation of adaptive immune responses through antigens bound to surface receptors of antigen presenting cells (APCs). T cell receptors (TCR) are responsible for initiating the immune response through their physical direct interaction with antigen-bound receptors on the APCs surface. The study of T cell interactions with antigens is considered of crucial importance for the comprehension of the role of immune responses in cancer growth and for the subsequent design of immunomodulating anticancer drugs. RNA sequencing experiments performed on T cells represented a major breakthrough for this branch of experimental molecular biology. Apart from the gene expression levels, the hypervariable CDR3α/β sequences of the TCR loops can now be easily determined and modelled in the three dimensions, being the portions of TCR mainly responsible for the interaction with APC receptors. The most direct experimental method for the investigation of antigens would be based on peptide libraries, but their huge combinatorial nature, size, cost, and the difficulty of experimental fine tuning makes this approach complicated time consuming, and costly. We have implemented in silico methodology with the aim of moving from CDR3α/β sequences to a library of potentially antigenic peptides that can be used in immunologically oriented experiments to study T cells' reactivity. To reduce the size of the library, we have verified the reproducibility of experimental benchmarks using the permutation of only six residues that can be considered representative of all ensembles of 20 natural amino acids. Such a simplified alphabet is able to correctly find the poses and chemical nature of original antigens within a small subset of ligands of potential interest. The newly generated library would have the advantage of leading to potentially antigenic ligands that would contribute to a better understanding of the chemical nature of TCR-antigen interactions. This step is crucial in the design of immunomodulators targeted towards T-cells response as well as in understanding the first principles of an immune response in several diseases, from cancer to autoimmune disorders.
Keywords: T-cell receptor (TCR); antigen recognition; ligand rational design; molecular mechanisms of adaptive immunity; receptor-peptide interaction.
Copyright © 2021 Iannuzzi, Rossetti, Spitaleri, Bonnal, Pagani and Mollica.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures



Similar articles
-
Immunobiological analysis of TCR single-chain transgenic mice reveals new possibilities for interaction between CDR3alpha and an antigenic peptide bound to MHC class I.J Immunol. 2001 Oct 15;167(8):4396-404. doi: 10.4049/jimmunol.167.8.4396. J Immunol. 2001. PMID: 11591764
-
Contribution of individual amino acids within MHC molecule or antigenic peptide to TCR ligand potency.J Immunol. 2000 Jan 15;164(2):861-71. doi: 10.4049/jimmunol.164.2.861. J Immunol. 2000. PMID: 10623833
-
Predicting CD4 T-cell epitopes based on antigen cleavage, MHCII presentation, and TCR recognition.PLoS One. 2018 Nov 6;13(11):e0206654. doi: 10.1371/journal.pone.0206654. eCollection 2018. PLoS One. 2018. PMID: 30399156 Free PMC article.
-
Thymic commitment of regulatory T cells is a pathway of TCR-dependent selection that isolates repertoires undergoing positive or negative selection.Curr Top Microbiol Immunol. 2005;293:43-71. doi: 10.1007/3-540-27702-1_3. Curr Top Microbiol Immunol. 2005. PMID: 15981475 Review.
-
Modification of human T-cell responses by altered peptide ligands: a new approach to antigen-specific modification.Intern Med. 1998 Oct;37(10):804-17. doi: 10.2169/internalmedicine.37.804. Intern Med. 1998. PMID: 9840700 Review.
References
-
- Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., et al. (2015). Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1 (2), 19–25. 10.1016/j.softx.2015.06.001 - DOI
-
- Berendsen H. J. C., Berendsen H. J. C., Van Der Spoel D., Van Drunen R. (1995). Gromacs: a message-passing parallel molecular dynamics implementation. Comp. Phys. Comm. 91, 43–56. 10.1016/0010-4655(95)00042-E - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous