

An Efficient Computational Model for Large-Scale Prediction of Protein-Protein Interactions Based on Accurate and Scalable Graph Embedding
- PMID: 33719344
- PMCID: PMC7953052
- DOI: 10.3389/fgene.2021.635451
An Efficient Computational Model for Large-Scale Prediction of Protein-Protein Interactions Based on Accurate and Scalable Graph Embedding
Abstract
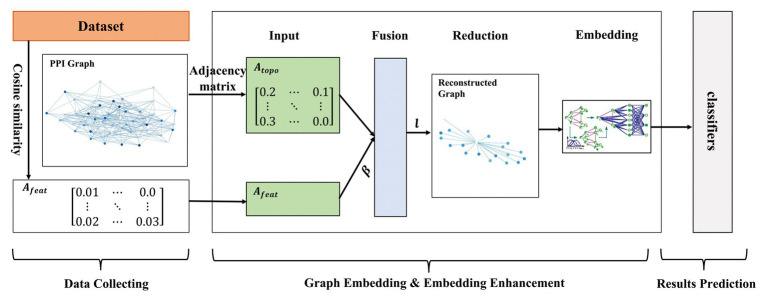
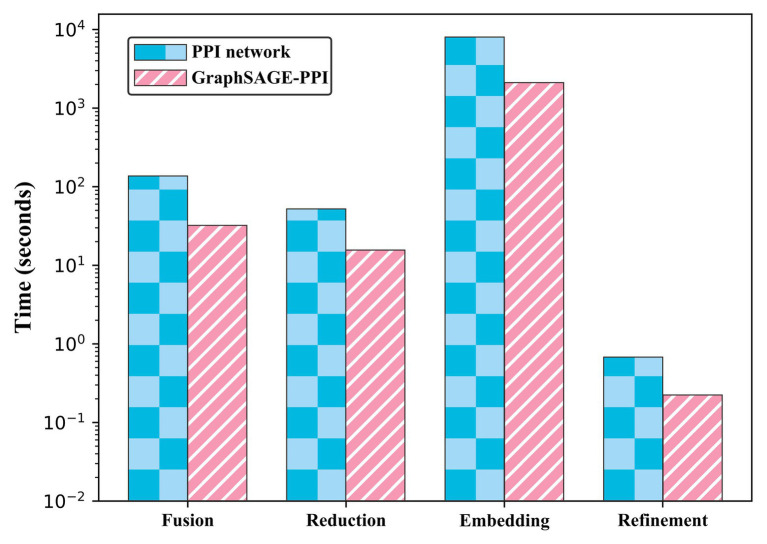
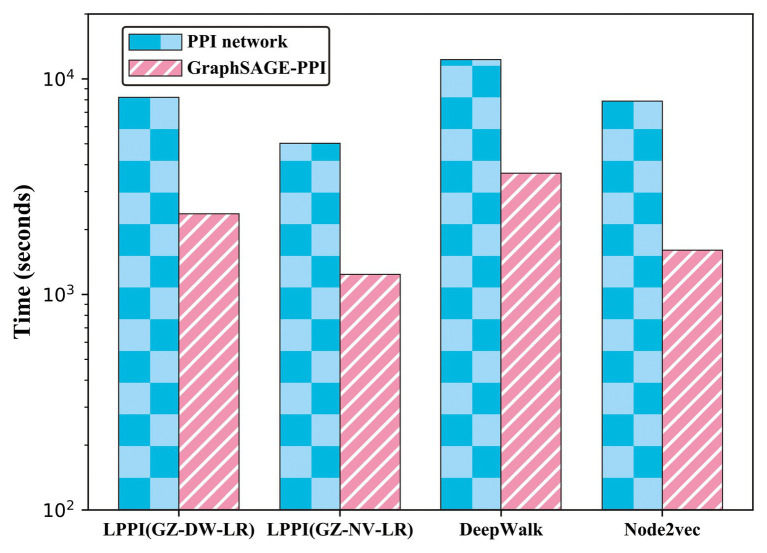
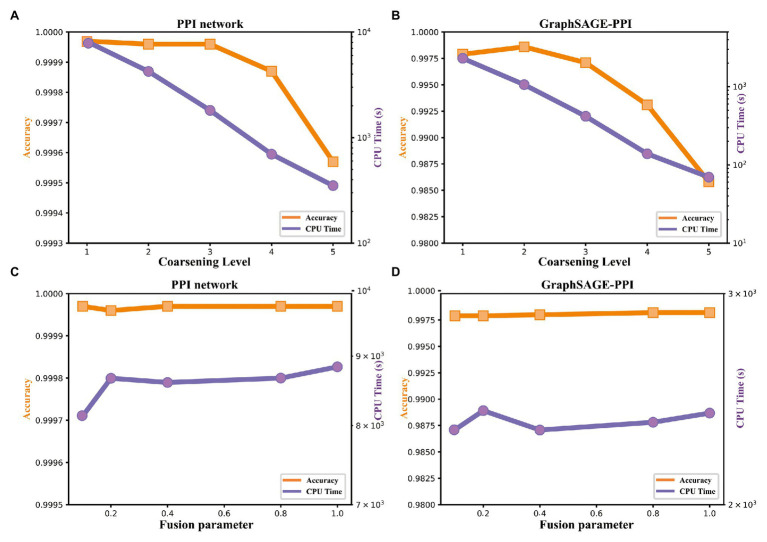
Protein-protein interaction (PPI) is the basis of the whole molecular mechanisms of living cells. Although traditional experiments are able to detect PPIs accurately, they often encounter high cost and require more time. As a result, computational methods have been used to predict PPIs to avoid these problems. Graph structure, as the important and pervasive data carriers, is considered as the most suitable structure to present biomedical entities and relationships. Although graph embedding is the most popular approach for graph representation learning, it usually suffers from high computational and space cost, especially in large-scale graphs. Therefore, developing a framework, which can accelerate graph embedding and improve the accuracy of embedding results, is important to large-scale PPIs prediction. In this paper, we propose a multi-level model LPPI to improve both the quality and speed of large-scale PPIs prediction. Firstly, protein basic information is collected as its attribute, including positional gene sets, motif gene sets, and immunological signatures. Secondly, we construct a weighted graph by using protein attributes to calculate node similarity. Then GraphZoom is used to accelerate the embedding process by reducing the size of the weighted graph. Next, graph embedding methods are used to learn graph topology features from the reconstructed graph. Finally, the linear Logistic Regression (LR) model is used to predict the probability of interactions of two proteins. LPPI achieved a high accuracy of 0.99997 and 0.9979 on the PPI network dataset and GraphSAGE-PPI dataset, respectively. Our further results show that the LPPI is promising for large-scale PPI prediction in both accuracy and efficiency, which is beneficial to other large-scale biomedical molecules interactions detection.
Keywords: GraphZoom; graph embedding; large-scale; protein-protein interaction; weighted graph.
Copyright © 2021 Su, You, Hu, Huang, Wang and Yi.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Graph embedding-based novel protein interaction prediction via higher-order graph convolutional network.PLoS One. 2020 Sep 24;15(9):e0238915. doi: 10.1371/journal.pone.0238915. eCollection 2020. PLoS One. 2020. PMID: 32970681 Free PMC article.
-
Graph-based prediction of Protein-protein interactions with attributed signed graph embedding.BMC Bioinformatics. 2020 Jul 21;21(1):323. doi: 10.1186/s12859-020-03646-8. BMC Bioinformatics. 2020. PMID: 32693790 Free PMC article.
-
DSSGNN-PPI: A Protein-Protein Interactions prediction model based on Double Structure and Sequence graph neural networks.Comput Biol Med. 2024 Jul;177:108669. doi: 10.1016/j.compbiomed.2024.108669. Epub 2024 May 29. Comput Biol Med. 2024. PMID: 38833802
-
Graph embedding and geometric deep learning relevance to network biology and structural chemistry.Front Artif Intell. 2023 Nov 16;6:1256352. doi: 10.3389/frai.2023.1256352. eCollection 2023. Front Artif Intell. 2023. PMID: 38035201 Free PMC article. Review.
-
Quantum Molecular Dynamics, Topological, Group Theoretical and Graph Theoretical Studies of Protein-Protein Interactions.Curr Top Med Chem. 2019;19(6):426-443. doi: 10.2174/1568026619666190304152704. Curr Top Med Chem. 2019. PMID: 30836919 Review.
Cited by
-
Protein Sequence Analysis landscape: A Systematic Review of Task Types, Databases, Datasets, Word Embeddings Methods, and Language Models.Database (Oxford). 2025 May 30;2025:baaf027. doi: 10.1093/database/baaf027. Database (Oxford). 2025. PMID: 40448683 Free PMC article.
-
A multi-source molecular network representation model for protein-protein interactions prediction.Sci Rep. 2024 Mar 14;14(1):6184. doi: 10.1038/s41598-024-56286-w. Sci Rep. 2024. PMID: 38485942 Free PMC article.
-
An Ensemble Classifiers for Improved Prediction of Native-Non-Native Protein-Protein Interaction.Int J Mol Sci. 2024 May 29;25(11):5957. doi: 10.3390/ijms25115957. Int J Mol Sci. 2024. PMID: 38892144 Free PMC article.
-
Multi-view heterogeneous molecular network representation learning for protein-protein interaction prediction.BMC Bioinformatics. 2022 Jun 16;23(1):234. doi: 10.1186/s12859-022-04766-z. BMC Bioinformatics. 2022. PMID: 35710342 Free PMC article.
-
Graph embedding on mass spectrometry- and sequencing-based biomedical data.BMC Bioinformatics. 2024 Jan 2;25(1):1. doi: 10.1186/s12859-023-05612-6. BMC Bioinformatics. 2024. PMID: 38166530 Free PMC article. Review.
References
-
- Belkin M., Niyogi P. (2003). Laplacian eigenmaps for dimensionality reduction and data. Neural Comput. 15, 1373–1396. 10.1162/089976603321780317 - DOI
-
- Deng C., Zhao Z., Wang Y., Zhang Z., Feng Z. (2019). ‘GraphZoom: a multi-level spectral approach for accurate and scalable graph embedding.’ Comput. Sci. [Preprint].
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials