

Very long chain fatty acid metabolism is required in acute myeloid leukemia
- PMID: 33720355
- PMCID: PMC8225921
- DOI: 10.1182/blood.2020008551
Very long chain fatty acid metabolism is required in acute myeloid leukemia
Abstract
Acute myeloid leukemia (AML) cells have an atypical metabolic phenotype characterized by increased mitochondrial mass, as well as a greater reliance on oxidative phosphorylation and fatty acid oxidation (FAO) for survival. To exploit this altered metabolism, we assessed publicly available databases to identify FAO enzyme overexpression. Very long chain acyl-CoA dehydrogenase (VLCAD; ACADVL) was found to be overexpressed and critical to leukemia cell mitochondrial metabolism. Genetic attenuation or pharmacological inhibition of VLCAD hindered mitochondrial respiration and FAO contribution to the tricarboxylic acid cycle, resulting in decreased viability, proliferation, clonogenic growth, and AML cell engraftment. Suppression of FAO at VLCAD triggered an increase in pyruvate dehydrogenase activity that was insufficient to increase glycolysis but resulted in adenosine triphosphate depletion and AML cell death, with no effect on normal hematopoietic cells. Together, these results demonstrate the importance of VLCAD in AML cell biology and highlight a novel metabolic vulnerability for this devastating disease.
© 2021 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: A.D.S. has received honoraria or consulting fees from Novartis, Jazz Pharmaceuticals, Otsuka, and Takeda Pharmaceuticals and research support from Medivir AB and Takeda. A.D.S. owns stock in AbbVie Pharmaceuticals. A.D.S. and P.A.S. are named on a patent application for the use of avocado compounds for the treatment of leukemia; however, this patent was not pursued passed the jurisdictional phase. P.A.S. has received honoraria from the Hass Avocado Board. The remaining authors declare no competing financial interests.
Figures



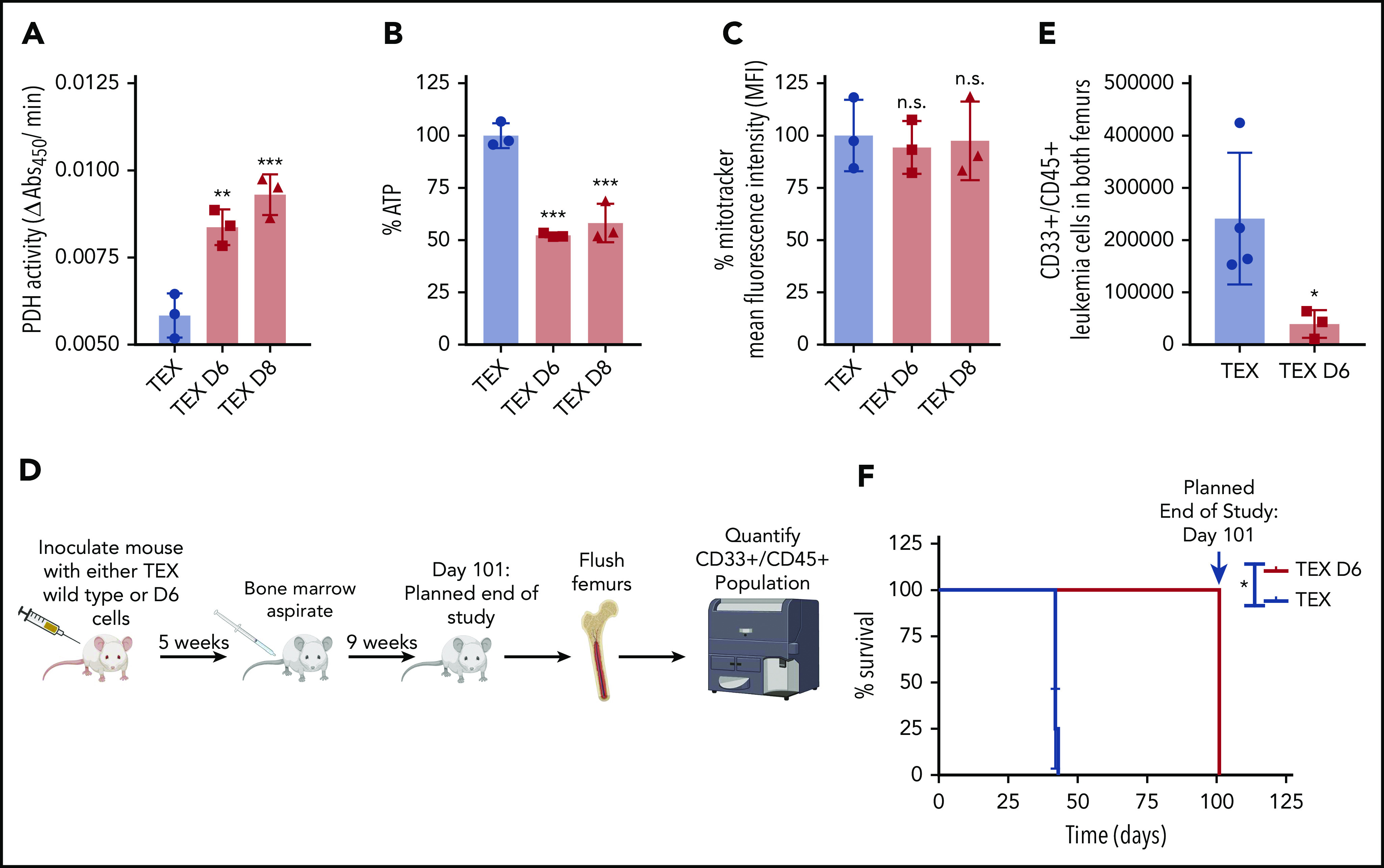



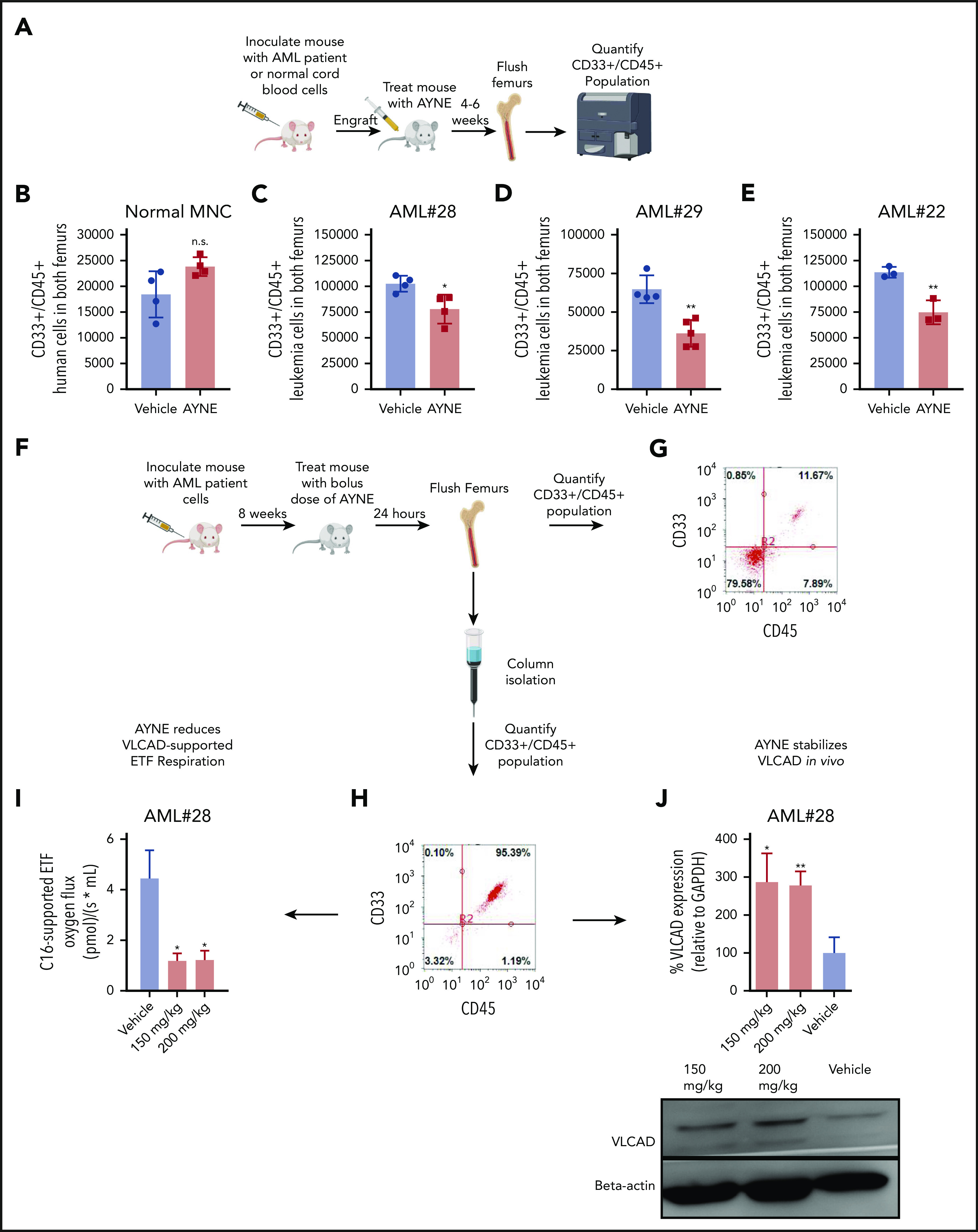
Comment in
-
Break the lifeline of AML cells.Blood. 2021 Jun 24;137(25):3465-3467. doi: 10.1182/blood.2021011475. Blood. 2021. PMID: 34165546 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
