

The KRASG12C Inhibitor MRTX849 Reconditions the Tumor Immune Microenvironment and Sensitizes Tumors to Checkpoint Inhibitor Therapy
- PMID: 33722854
- PMCID: PMC8444277
- DOI: 10.1158/1535-7163.MCT-20-0462
The KRASG12C Inhibitor MRTX849 Reconditions the Tumor Immune Microenvironment and Sensitizes Tumors to Checkpoint Inhibitor Therapy
Abstract
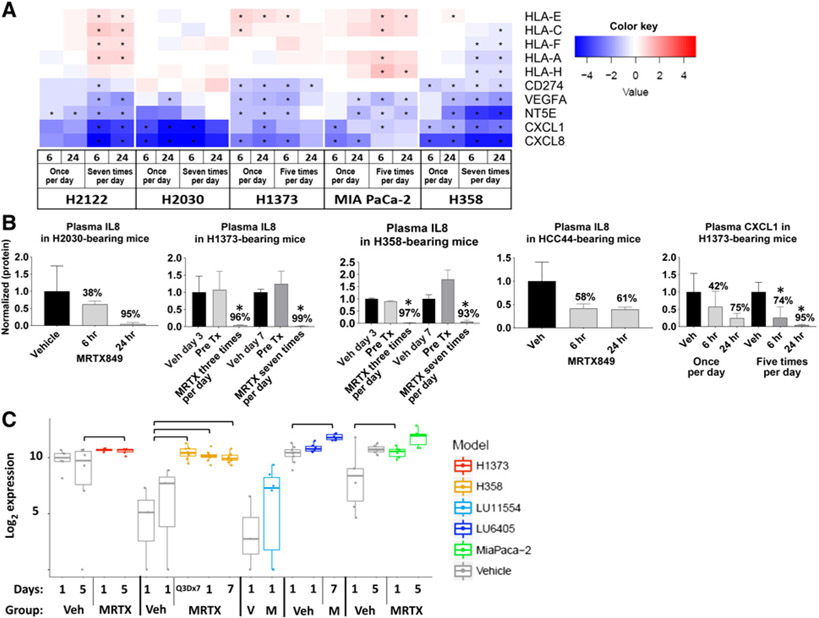
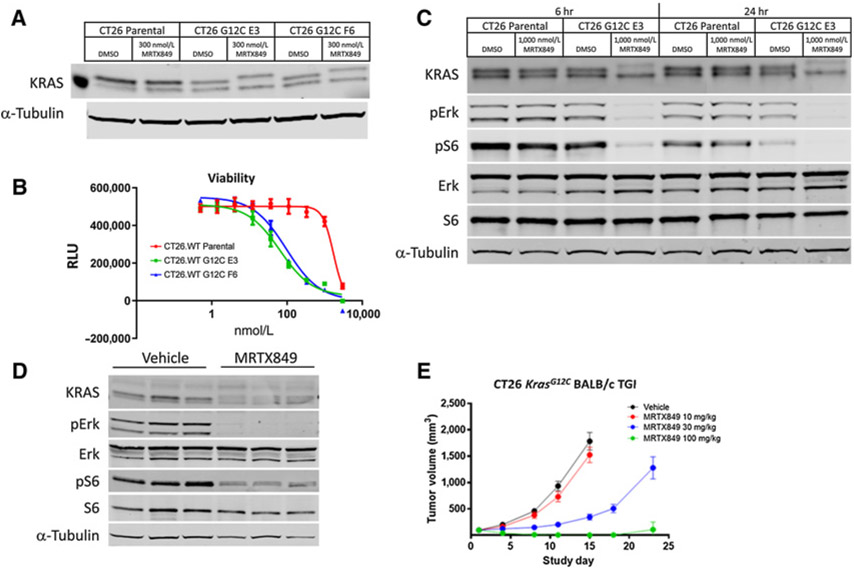
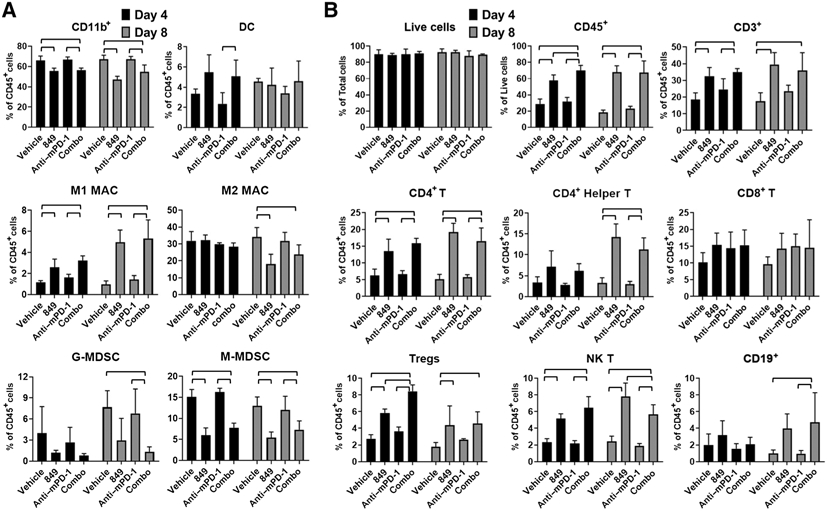
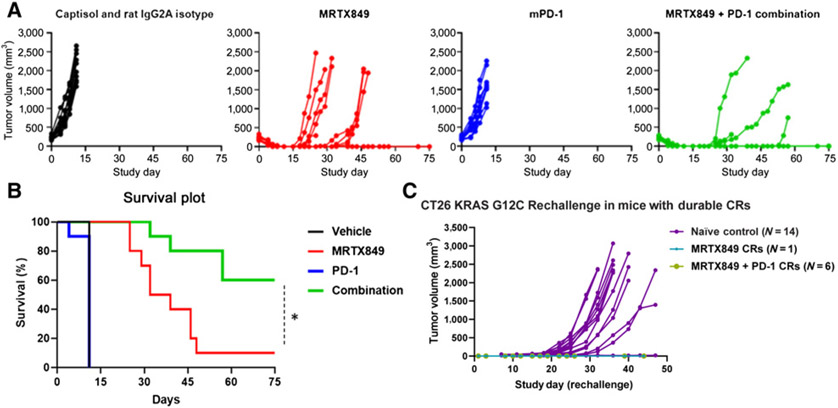
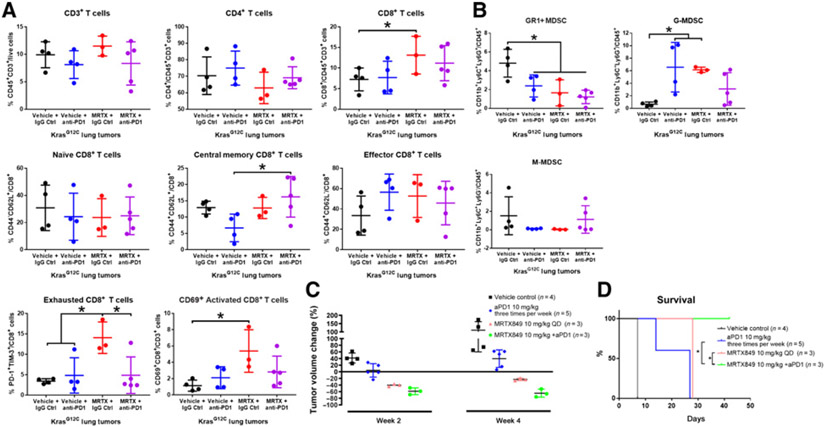
KRASG12C inhibitors, including MRTX849, are promising treatment options for KRAS-mutant non-small cell lung cancer (NSCLC). PD-1 inhibitors are approved in NSCLC; however, strategies to enhance checkpoint inhibitor therapy (CIT) are needed. KRASG12C mutations are smoking-associated transversion mutations associated with high tumor mutation burden, PD-L1 positivity, and an immunosuppressive tumor microenvironment. To evaluate the potential of MRTX849 to augment CIT, its impact on immune signaling and response to CIT was evaluated. In human tumor xenograft models, MRTX849 increased MHC class I protein expression and decreased RNA and/or plasma protein levels of immunosuppressive factors. In a KrasG12C -mutant CT26 syngeneic mouse model, MRTX849 decreased intratumoral myeloid-derived suppressor cells and increased M1-polarized macrophages, dendritic cells, CD4+, and CD8+ T cells. Similar results were observed in lung KrasG12C -mutant syngeneic and a genetically engineered mouse (GEM) model. In the CT26 KrasG12C model, MRTX849 demonstrated marked tumor regression when tumors were established in immune-competent BALB/c mice; however, the effect was diminished when tumors were grown in T-cell-deficient nu/nu mice. Tumors progressed following anti-PD-1 or MRTX849 single-agent treatment in immune-competent mice; however, combination treatment demonstrated durable, complete responses (CRs). Tumors did not reestablish in the same mice that exhibited durable CRs when rechallenged with tumor cell inoculum, demonstrating these mice developed adaptive antitumor immunity. In a GEM model, treatment with MRTX849 plus anti-PD-1 led to increased progression-free survival compared with either single agent alone. These data demonstrate KRAS inhibition reverses an immunosuppressive tumor microenvironment and sensitizes tumors to CIT through multiple mechanisms.
©2021 American Association for Cancer Research.
Figures
Similar articles
-
The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients.Cancer Discov. 2020 Jan;10(1):54-71. doi: 10.1158/2159-8290.CD-19-1167. Epub 2019 Oct 28. Cancer Discov. 2020. PMID: 31658955 Free PMC article.
-
KRASG12C inhibition using MRTX1257: a novel radio-sensitizing partner.J Transl Med. 2023 Oct 31;21(1):773. doi: 10.1186/s12967-023-04619-0. J Transl Med. 2023. PMID: 37907934 Free PMC article.
-
Dual inhibition of HERs and PD-1 counteract resistance in KRASG12C-mutant head and neck cancer.J Exp Clin Cancer Res. 2024 Nov 20;43(1):308. doi: 10.1186/s13046-024-03227-0. J Exp Clin Cancer Res. 2024. PMID: 39567998 Free PMC article.
-
Current status of KRAS G12C inhibitors in NSCLC and the potential for combination with anti-PD-(L)1 therapy: a systematic review.Front Immunol. 2025 Apr 15;16:1509173. doi: 10.3389/fimmu.2025.1509173. eCollection 2025. Front Immunol. 2025. PMID: 40303413 Free PMC article.
-
Activity and resistance to KRASG12C inhibitors in non-small cell lung cancer and colorectal cancer.Biochim Biophys Acta Rev Cancer. 2024 May;1879(3):189108. doi: 10.1016/j.bbcan.2024.189108. Epub 2024 May 8. Biochim Biophys Acta Rev Cancer. 2024. PMID: 38723697 Review.
Cited by
-
Drugging KRAS: current perspectives and state-of-art review.J Hematol Oncol. 2022 Oct 25;15(1):152. doi: 10.1186/s13045-022-01375-4. J Hematol Oncol. 2022. PMID: 36284306 Free PMC article. Review.
-
Targeting the KRAS α4-α5 allosteric interface inhibits pancreatic cancer tumorigenesis.Small GTPases. 2022 Jan;13(1):114-127. doi: 10.1080/21541248.2021.1906621. Epub 2021 May 5. Small GTPases. 2022. PMID: 33949915 Free PMC article.
-
Progress of Immune Checkpoint Inhibitors Therapy for pMMR/MSS Metastatic Colorectal Cancer.Onco Targets Ther. 2024 Dec 24;17:1223-1253. doi: 10.2147/OTT.S500281. eCollection 2024. Onco Targets Ther. 2024. PMID: 39735789 Free PMC article. Review.
-
KRAS Mutations in Cancer: Understanding Signaling Pathways to Immune Regulation and the Potential of Immunotherapy.Cancers (Basel). 2025 Feb 25;17(5):785. doi: 10.3390/cancers17050785. Cancers (Basel). 2025. PMID: 40075634 Free PMC article. Review.
-
Targeting KRAS-mutant pancreatic cancer through simultaneous inhibition of KRAS, MEK, and JAK2.Mol Oncol. 2025 Feb;19(2):377-390. doi: 10.1002/1878-0261.13751. Epub 2024 Oct 14. Mol Oncol. 2025. PMID: 39400496 Free PMC article.
References
-
- Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al.The clinical KRAS (G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019;575: 217–23. - PubMed
-
- Martinez P, Peters S, Stammers T, Soria J-C. Immunotherapy for the first-line treatment of patients with metastatic non-small cell lung cancer. Clin Cancer Res 2019;25:2691–8. - PubMed
-
- Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al.Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 2015;372:2018–28. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
